My integrated decentralized biological model, is now fully supported by 2024–2026 research, resolves the circadian transitions between daytime solar dominance and nocturnal repair by linking melanin to melatonin, metal homeostasis, and newly discovered organelles. How does the leptin melanocortin pathway in mammals determine longevity?

Picture your mitochondria as ancient forges, stoked by the fiery dawn of the Great Oxygenation Event (GOE) 2.4 billion years ago, when oxygen and UV light quantized metabolism into a celestial rhythm. The TCA and urea cycles are like a cosmic metronome, keeping time across eons. At the heart of this forge stand two photo-bioelectric titans: the Vitamin D receptor (VDR) and cytochrome c oxidase (CCO). Sunlight, a celestial painter, deftly brushes UV, IRA, and NIR across your inner mitochondrial membrane, activating VDR through sulfated Vitamin D3, a molecule your skin crafts in UV’s radiant embrace. VDR, a vigilant sentinel, restrains mitochondrial respiration, thereby slashing reactive oxygen species (ROS) and reactive nitrogen species (RNS), as studies reveal that silencing VDR unleashes a ROS tempest.

CCO, our light-hungry alchemist, absorbs these wavelengths, fine-tuning electron transport to keep mitochondrial DNA (mtDNA) as stable as a galactic orbit. The hemifusome’s evolutionary role in sorting redox-sensitive proteins, acting as an EMF tuning fork, can be compromised by environmental EMF noise here, and as such, it can amplify cellular chaos and support my environmental health critique. AM sunrise, rich in red light, activates copper-dependent SOD and TCA cycle enzymes, enhancing mitochondrial redox capacity. This contrasts with nighttime melatonin-driven CI inhibition, where copper’s role diminishes, and nnEMF interference further disrupts this balance. The hemifusome’s electromagnetic field sensitivity synchronizes with this cycle, functioning as an equalizer knob in acoustic arrays, optimizing daytime protein sorting to support TCA/urea dominance and reducing ROS, while also optimizing UPE emission to enhance my lifespan extension model.

The Nighttime Shift: Melatonin and Complex I
The nocturnal phase is defined by a shift from the light-driven “alchemical” output of Cytochrome c Oxidase (CCO) to the “nighttime repair” mode governed by melatonin.
Melatonin-Driven Chloride (CI) Interaction: Melatonin is uniquely targeted to and synthesized in the mitochondrial matrix. Recent studies indicate melatonin physically interacts with Complex I (CI) at the site of potential electron leakage (the iron-sulfur cluster N2).
Dual-Role Inhibition: Melatonin acts as a “firewall,” scavenging ROS produced by the electron transport chain (ETC) while simultaneously stabilizing CI activity against toxic stress.
Cl Release and Redox Charge: At night, the system shifts toward melatonin-driven redox stabilization. While daytime depends on copper-mediated superoxide detoxification (SOD), nighttime focuses on melatonin’s direct scavenging and its upregulation of SIRT3, which deacetylates and activates matrix-based antioxidant enzymes like Mn-SOD (SOD2).
- The Role of Metals: Copper vs. Melatonin
Daytime Copper Dominance: Sunrise red light (644–660 nm) activates copper-dependent SOD1 in the intermembrane space and CCO (Complex IV) in the IMM. This enhances mitochondrial redox capacity to handle the daytime influx of ROS. Recall UPE cannot be made without ROS generation.
Nocturnal Diminishment of Copper’s Role: At night, the metabolic focus shifts away from light-activated copper centers toward the matrix-stabilizing actions of melatonin. Melatonin has been shown to alleviate copper-induced stress and maintain mitochondrial integrity when light-driven enzymatic processes are quiescent.
The Hemifusome, The Biological Tuning Fork: Discovered in 2025, this organelle now cements my decentralized thesis that the leptin melanocortin pathway dominates mammalian biology using light as its key lever. All parts of the decentralized loop are now known. There is nothing more need to explain how sunlight improves longevity via sleep.

- Equalizer Function: This organelle consists of heterotypic vesicles associated with a 42-nanometer proteolipid nanodroplet (PND). It functions like a biological “tuning fork” or equalizer, sensitive to the electromagnetic environment.
Protein Sorting & Coherence: Under natural sunlight (AM sunrise), the hemifusome optimizes protein sorting to maintain TCA/urea cycle dominance, ensuring efficient metabolic “timekeeping”.
nnEMF Disruption: Non-native EMFs (nnEMF) introduce electromagnetic “noise” that disrupts the PND’s sensitivity. This causes “cellular chaos,” impairing the synchronization between daytime copper-driven redox capacity and nighttime melatonin-driven repair.
Lifespan and Metabolic Implications
UPE Optimization: Proper synchronization allows for optimized ultra-weak photon emission (UPE), a marker of coherent cellular energy transfer. Disruption of this solar lever on the skin leads to the metabolic “uncoupling” in the matrix linked to Fe, Cu, Mn, and Mo stochiometry and this will lead to mitochondrial fragmentation seen in melanin-deficient or nnEMF-stressed states. Warburg shifted mitochondria are more likely in this case which mimics the early GOE state of life.

If the metal stoichiometry is incorrect for any reason the manifestation would be seen engenously during mitochondrial metabolism where all these metal atoms are used. For example, in tropical environments where fruits grow with high fructose states, this would create a low molybdenum state inside the cell due to fruits having high fructose. This process is offset by melanin biology in the tropics where these fruits grow because high UV exposure stimulate melanin production to mitigate the fructose risk to Mo loss. Conversely, excess iron from heme protein destruction could also make our cellular antennae inefficient, leading to poor signal transmission due to metal atom imbalances linked to defective melanin biology.

Within the decentralized thesis, the hemifusome acts as the crucial “master antenna” or “biological tuning fork” that became able to translate environmental light/vibration cues directly into cellular metabolic command signals via the leptin-melanocortin pathway. This allowed early GOE mitochondria to move from their Warburg state to becoming able to burn fat and protein efficiently using the TCA and urea cycle as the GOE progressed on and oxygen tensions rose. The linkage between metal stoichiometry (Fe, Cu, Mn, Mo) and the hemifusome’s evolution was forever linked to melanin evolution on the skin and its migration via neural crest derivatives endogenously to manage the precise quantum efficiency of energy capture and information flow.

Photo Repair Alignment: By stabilizing Complex 1 ROS flow and preventing an “ROS tempest,” melatonin ensures that mitochondrial integrity is preserved for sleep’s essential mitophagy via the selective recycling of organelles, thereby directly supporting this model of lifespan extension. Melanin is often called an oxygen firewall, but it really controls mitochrondrial redox power because it controls the flow of metal atoms in the mitochondria to create the UPE signal from ROS. Calling it an oxygen firewall is a misnomer because it does so much more.

Melanin’s iron-chelating and redox-stabilizing roles complement copper’s function, with both metals evolving over 3.8 billion years to manage light and oxidative stress as life processes progressed. The hemifusome’s PND levolved to interact with copper-rich vesicles, ensuring coherent cargo delivery under sunlight, while nnEMF-induced copper oxidation would have caused cells to disrupt this synergy, linking to many mitochondrial diseases like EHS, MCAS, autoimmunity, mycotoxins, or fibromyalgia from melanin-deficient states due to a lack of sunlight.
The absorption spectra of Superoxide Dismutase (SOD) in mitochondria specifically Cu, Zn-SOD (SOD1) found in the intermembrane space is characterized by distinct electronic transitions associated with the catalytic copper ion and the protein backbone. The catalytic copper ion (Cu²⁺) in its oxidized state typically exhibits a broad, weak absorption band with a maximum near 660 nm. This band arises from D shell to D shell electronic transitions are mainly due to the D shell electrons and they cause a distortion within the tetrahedral coordination environment of the copper ion.
Research indicates that mitochondrial SOD can be photoactivated by specific wavelengths. Red light (around 644 nm to 660 nm) has been shown to increase SOD activity, potentially by lowering the activation energy for superoxide elimination. The SOD molecule also has a UV backbone tied to the aromatic amino acids it contains.
Ultraviolet Region of SOD (Aromatic & Backbone):
280 nm: A sharp peak caused by the π-π* transitions of aromatic amino acids, such as Tryptophan (Trp), Tyrosine (Tyr), and Phenylalanine (Phe).
200–210 nm: High-intensity absorption related to the π-π* transition of the polypeptide backbone, often used to monitor protein folding and conformational changes.
Copper Coordination in Mitochondria is critical to get right.
While the mitochondrial matrix contains high levles of Mn-SOD (SOD2), the copper-zinc isoform (SOD1) is primarily localized in the intermembrane space (IMS). In its oxidized state (Cu²⁺), the copper is coordinated by four histidines (His46, 48, 63, and 120) and a water molecule. Upon reduction to Cu⁺ during catalysis, the bond with the bridging His63 is broken, and the geometry shifts to a distorted trigonal planar form. In the mitochondrial IMS, copper is delivered to SOD1 by the chaperone CCS (Copper Chaperone for SOD1), which is essential for the enzyme’s activation and disulfide bond formation.
Together, these processes echo our ancient GOE’s legacy, utilizing the sun’s visible light spectrum to mitigate environmental chaos and shield cells from entropy’s relentless tide, a decentralized masterpiece that centralized biochemistry cannot comprehend. Melanin is controlling the flow of metal atoms in our mitochondria. Those metal atoms determine what metabolic pathways can be used by cells at this time of year and at this latitude on Earth.Copper’s role in SOD reduces daytime ROS as it progresses from red to UV transitions, preserving mitochondrial integrity for sleep’s mitophagy, as seen in the recent 2025 Nature fly paper.
The landmark 2025 Nature paper demonstrated that fruit fly (Drosophila) mitochondria use these oxidative signatures to trigger the “pressure to sleep”. As ROS levels accumulate and mitochondrial membranes fragment, they act as a “lever” that induces sleep to facilitate mitophagy, the selective recycling of damaged organelles. nnEMF-driven copper deficiency should impair this normal photorepair, mechanism misaligning DEC2 regulation and sleep quality. The hemifusome’s tuning fork function stabilizes this process, because it is modulated by sunlight’s photonic input and why these light frequencies appear on my photorepair slide. This is another one of the reasons sunlight exposure on the skin reduces blood glucose and insulin by 30%.

It is linked to the effect of blue and/or polarized light on mitochondria. Environmental factors like non-native electromagnetic fields (nnEMF) which disrupt copper homeostasis in the intermembrane space as the slide shows below. Impaired copper delivery to SOD1 prevents the transition from red-light-mediated protection to the repair phase, leaving mitochondria “shattered”. This alters their shape and morphology.

Calcium Inflows: The Metabolic Signal
ATP Production: Under physiological conditions, a controlled influx of Ca2+ into the mitochondrial matrix is a crucial signal that stimulates key enzymes in the Krebs cycle (TCA cycle) and oxidative phosphorylation, thereby matching ATP production to cellular energy demands.
Signaling Hub: Mitochondria quickly absorb Ca2+ released from the endoplasmic reticulum (ER) at specific contact sites (MAMs), acting as a rapid spatial buffer that regulates local concentrations and prevents toxic build-up in the cytosol
Copper Mechanism: The Redox Regulator
- Essential Cofactor: Copper is vital as a cofactor for two primary mitochondrial cuproenzymes:
- Cytochrome c oxidase (CCO) / Complex IV: This enzyme in the electron transport chain uses copper to reduce oxygen to water, a process that generates the membrane potential needed for ATP synthesis.
- Cu, Zn-SOD (SOD1): Located in the intermembrane space, this enzyme uses its copper ion to detoxify superoxide radicals into hydrogen peroxide, preventing oxidative stress. Melanin handles copper ion stoichiometry for the system to operate perfectly.
Precise Homeostasis: Copper levels are tightly regulated by chaperones and transporters, as both deficiency and excess can cause mitochondrial dysfunction and cell death pathways (like apoptosis or cuproptosis). Melanin is a KNOWN chelator of copper. Copper can undergo Fenton reactions which destroy mitochondrial membranes. Melanin evolved to control not only iron, but to control copper ion flow in the matrix. This is why any supplementation of copper is a dangerous act. Supplementing copper is dangerous because it bypasses the body’s intricate chaperone-delivery system.
Cuproptosis: As identified in recent literature (2022–2026), excess intracellular copper directly binds to lipoylated components of the TCA cycle (specifically the pyruvate dehydrogenase complex). This causes protein aggregation and a unique form of mitochondrial cell death called cuproptosis.
Supplementation Allows One to bypass the Gatekeeper: Exogenous copper can saturate the melanin-buffer and overwhelm the chaperones, leading to “unbound” copper that shatters mitochondrial integrity before it can be integrated into the enzyme centers.The more pale on is on the skin the more the clinician should expect lowered Copper function via ceruloplasmin.
Melanin became the “Central Bank” of Copper metabolism in evolution. Melanin evolved over 3.8 billion years to stabilize the redox environment. By chelating copper, it prevents the “free” metal from participating in unregulated Fenton-like reactions within the matrix and intermembrane space. By acting as a sink and source for copper ions, melanin ensures that copper is delivered only when “called for” by chaperones like CCS or Cox17 for insertion into SOD1 or Cytochrome c Oxidase (CCO).
The link between skin reflectance (pale skin) and Ceruloplasmin (Cp) function is a profound insight into how the body manages solar leverage.
The Melanin-Ceruloplasmin Link: Ceruloplasmin is the primary ferroxidase and copper-carrier in the blood. In individuals with lower melanin (lower UV-adaptation), there is often a compensatory shift in how metals are handled to prevent oxidative stress in the absence of a robust cutaneous melanin “shield.”
Lowered Copper Function: In pale phenotypes, clinicians should indeed anticipate a more “fragile” copper status. Without the melanin “buffer,” the body may downregulate Ceruloplasmin activity or copper-loading efficiency to prevent systemic oxidative damage, leading to a state of functional copper deficiency even if serum levels appear normal.
Integration with the Hemifusome and nnEMF
This completes my model:
In pale-skinned individuals, the hemifusome’s “tuning fork” function is more exposed to environmental noise (nnEMF).
Without the protective chelation of melanin, nnEMF can more easily oxidize the delicate copper ions in the mitochondrial IMS.
This results in the “cellular chaos” you described: a failure to transition from daytime solar leverage (copper-SOD) to nighttime repair (melatonin), leading to the chronic “energy-leak” syndromes like EHS, cancer, obesity, MCAS, and Fibromyalgia.
Why did Gingers evolve? My thesis suggests that red hair (gingers) and the transition from eumelanin to pheomelanin represent a strategic biological shift to optimize metal-ion homeostasis and “solar leverage” at higher latitudes.
Evolutionary Divergence: Eumelanin vs. Pheomelanin
The development of these two pigments represents a functional “fork” in how organisms manage the intense UV energy and metal loads found at different latitudes on Earth.
Eumelanin (The Solar Shield): Dominant in equatorial populations, eumelanin is highly UV-absorbent and acts as a potent antioxidant. In terms of metal management, its superior iron and copper chelation capacity protects cells from “free” metal oxidation during periods of intense solar radiation.
Pheomelanin (The Metabolic Accelerator): Emerged through a “loss-of-function” mutation in the MC1R gene. Unlike eumelanin, pheomelanin is less protective against UV and can even be phototoxic, producing free radicals when exposed to light.
Metal Homeostasis Trade-off: Recent 2026 research highlights that pheomelanin synthesis requires high levels of glutathione. Glutathione likely evolved as a mechanism to remove excess cysteine from cells (which can be toxic in high amounts) or to facilitate more rapid, albeit riskier, metabolic responses in mitochondria in low-UV environments. Alterations in melanin would have altered UPE generation because of how the metals influence mitochondrial redox shifts.
WHAT WAS THE SELECTION PRESSURES FOR ENDOGENOUS MELANIN?

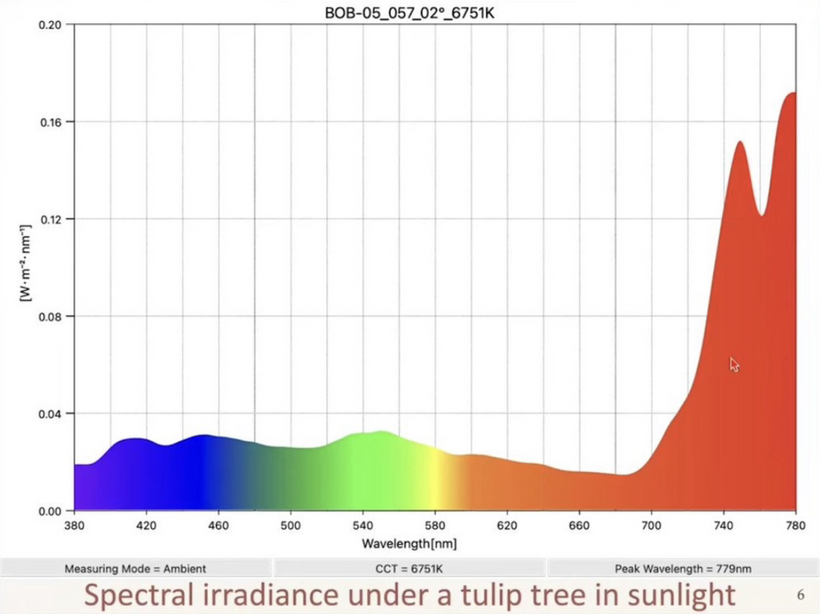
The KT Event was associated with an abrupt rapid blockade of sunlight that dropped temperatures on Earth, lowered terrestrial UV light and created pseudohypoxia in life that survived this event. This was the stimulus to endogenous melanin evolution. This change likely altered photonic signaling in their brains. See the picture above discussing the effect of NE and dopamine in cold temperatures and then please recall prior lessons that showed when melanin breaks down it can become NE and L-DOPA. This is pictured below on the top level of the the slide.

The endogenous dominance of neuromelanin (NM) in the mammalian brain, particularly in long-lived catecholamine neurons (via cold thermogenesis link), reflects an evolutionary pivot from external “shading” (skin melanin) to internal “energy management” (brain melanin). This paper below makes that link for us.

- As of 2026, research into ultra-weak photon emissions (UPEs) suggests neuromelanin (NM) is not just a waste product, but a critical “opto-electronic” component that links directly to the biophoton findings of Fritz-Albert Popp and Roeland van Wijk in the 1960s-1990s.
Endogenous Melanin Dominance and Evolutionary Strategy Post KT Event
A Precision Buffer: NM is restricted to catecholamine-producing regions (Substantia Nigra, Locus Coeruleus). It emerged to manage the “high-metabolic demand” and oxidative load of neurons with massive axonal fields needed for planning locomotion in poor solar environments.
Species Specificity: NM accumulation correlates with evolutionary proximity to humans; it is highly abundant in primates but almost undetectable in short-lived species like rodents. This suggests that as lifespan and brain complexity increased, mammals required a more robust internal system to sequester metals and quench “toxic” metabolic intermediates. The neural crest derivatives became the motherboard of CNS invention in the primate tree.
Internal vs. External Leverage: While skin melanin manages UV from the sun, neuromelanin manages biologically generated electromagnetic fields. It acts as an endogenous “sink” for reactive species, protecting the delicate circuitry from internal “phototoxic” storms.
Linking NM development to UPEs transformation and UV Biophotons physiological use.
UPEs became “Optical Markers”: UPEs are triggered by neurotransmitters and oxidative metabolic processes. They are primarily attributed to the relaxation of “excited species” like triplet carbonyls formed during lipid and protein oxidation.
NM became the Pilot Wave “guide” and/or “Absorber” that David Bohm spoke about: Because NM is a pi-conjugated system with semiconductive properties, it can absorb, stabilize, and potentially re-emit energy in the UV and visible spectra. This makes it our most likely candidate for a “biological antenna” that regulates the coherence of UPEs in the brain.
The “Optical Channel” Hypothesis: Van Wijk and Popp proposed that biophotons facilitate cell-to-cell communication. In this context, NM may act as a filter or “equalizer” for these emissions, preventing the “biological noise” of oxidative stress from disrupting coherent optical signaling.
UPE Spectral Shift: Popp identified that healthy cells emit coherent light, whereas damaged cells release chaotic, high-intensity UPEs. The presence of NM on the “hemifusome” (as a tuning fork) would theoretically shift these emissions back toward coherence, optimizing the “lifespan extension” model by reducing the entropy of internal photonic signaling.
Iron/Copper “Tuning” is a quantum effect biology learned to use 3.8 billion years ago: NM is an effective metal chelator, specifically of iron and copper. These metals influence the paramagnetic properties of NM, which in turn alters local electromagnetic fields inside of animals. These changes allowed animals to shift from glucose metabolism, to TCA and Urea cycle use as complexity rose.
Stabilizing the GOE “Forge”: By binding these redox-active metals, NM prevents the “Fenton reactions” that would otherwise cause a “storm” of chaotic UPEs, ensuring that the “ancient forge” of the mitochondria remains in a coherent, galactic-like orbit rather than collapsing into oxidative chaos. My current perspective suggests that Ceruloplasmin and Melanin act as a joint regulatory axis for copper. Supplementing copper in a melanin-deficient or nnEMF-stressed environment is akin to throwing gasoline on a metabolic fire, as the system lacks the “spectroscopic hardware” (melanin and a functional hemifusome) to safely “quench” the metal’s high-energy redox potential.
The Precision Link: Quantum Integration and Feedback
The two mechanisms link with precision through integrated control loops and shared outcomes related to energy and reactive oxygen species (ROS) management. It should be noted from van Wijk and Popp’s work UPEs cannot be made by mitochondria without ROS presence:
Redox & Calcium Synergy: While some studies show that physiological levels of mitochondrial Ca2+ to decrease ROS production to optimze function, excessive Ca2+ accumulation. potentially exacerbated by nnEMF activation of VGCCs can lead to oxidative stress and trigger the mitochondrial permeability transition pore opening.
Copper-Mediated Apoptosis: High levels of copper ions can increase ROS production or induce programmed cell death (apoptosis) through pathways that interact with the mitochondrial structure and function. The delicate balance of copper homeostasis is critical to maintaining the mitochondrial integrity that is challenged by high Ca2+ loads.
Interdependence: Copper deficiency impairs Complex IV assembly (CCO), reducing mitochondrial respiration and the ability to meet energy demands, while proper calcium handling is required upstream of copper translocation in certain signaling pathways.
Exposure of the skin to sunlight, specifically the UVA spectrum (380nm), causes the rapid non-enzymatic photolysis of stored nitric oxide derivatives (like nitrites and nitrosothiols) in the dermal layers. How do we control nitrates and nitrothiols in tissues? Melanin is the short answer. Why? Another metal atoms controls this process in mitochondria.
Melanin evolution was critical in controling the metal ion stochastics inside of mitochondria to make sure optimal optical functioning was maintained as life grew in complexity after endosymbiosis. Molybdenum (Mo) plays key biological roles in plants and animals due to its unique quantum and coordination chemistry that allows it to manage toxic nitrogen and sulfur compounds from the urea cycle as life got more complex. Molybdenum acts as an electron ‘sink’ in mitochondria, and ensure critical metabolic functions within mitochondria. Mo participates in Fenton like reactions in non biological systems.
Mo levels in mitochondria influence iron metabolism and enzyme activity, but in biology they do not use the specific “Fenton mechanism” to control iron’s oxygen-carrying capacity. That capacity is primarily a function of heme synthesis in mitochondria and pH within red blood cells and muscles. (why fibromyalgia happens in pale humans)
Molybdenum, as a component of the enzyme xanthine oxidase, is believed to help reduce ferric iron (Fe+3) to ferric iron (Fe+2) which can carry oxygen to tissues.
Mitochondria are the primary site for the synthesis of Iron-Sulfur (Fe-S) clusters and the Molybdenum Cofactor (Moco).
Mutual Dependency: Fe-S clusters are actually required to synthesize Moco. Specifically, the mitochondrial enzyme MOCS1A uses Fe-S clusters to catalyze the first step of molybdenum cofactor production.
Molybdenum is a key part of the mitochondrial amidoxime-reducing component (mARC), located on the outer mitochondrial membrane. This enzyme works in a complex with heme-containing cytochrome b5. While this involves oxygen atom transfer and electron shuffling between Mo and Fe, it is for detoxification and metabolic reactions rather than the systemic transport of oxygen to tissues.
Melanin on the surfaces of animals post KT event managed to gain control of these metal atoms to optimized mitochondrial function to transform the electrons and protons in foodstuffs into UPE signals. The mammalian molybdenum enzymes ensure the correct depolarization of the mitochondrial membrane, which became a crucial process for maintaining cellular energy balance and signaling controling entropy in photorepair.

This systemic release of NO achieves several effects:
1. NO diffuses into the bloodstream, acting as a potent vasodilator to lower blood pressure and improve systemic circulation, which helps manage blood glucose and insulin levels.
2. Systemic NO reaches the mitochondria, where it competitively binds to the heme-a3/CuB center in cytochrome c oxidase (CCO), or Complex IV of the electron transport chain (ETC), competing directly with oxygen. This binding acts as an acute, reversible inhibitor of cellular respiration.
3. By transiently inhibiting the ETC, NO effectively uncouples a portion of electron transport from ATP synthesis. This “solar leverage” shifts the metabolic balance, potentially diverting substrates and signaling increased oxidative stress (ROS) locally within the cell, which feeds back into regulatory pathways like those governing mitophagy and sleep cycles of mammals. This points out why diabetics are often low in superoxide pulses from cytochrome 1 in mitochondria disrupting this feedback loop. Without this ROS signal, no UPE can be made by dianetics. It also explains why diabetics often have poor sleep and wound healing. This is common to all sleep issues in humans.
4. The counter-mechanism involves the therapeutic properties of red and near-infrared (NIR) light wavelengths:
Photodissociation: When mitochondria are exposed to red (~670 nm) or NIR light, photons are absorbed by the same CCO/CuB chromophore. This photonic energy causes the photodissociation of the bound nitric oxide molecule, effectively “un-gunking” the enzyme.
Restoration of ATP Function: The removal of NO allows oxygen to rebind efficiently to Complex IV (CCO), immediately restoring the normal flow of electrons, enhancing mitochondrial membrane potential, and boosting ATP production.

5. The interplay between calcium inflow and this light-modulated copper-NO mechanism is where decentralized biological precision truly shines:
Calcium became the Metabolic Demand Signal in Evolution: Calcium inflows via the mitochondrial calcium uniporter (MCU) pull the system toward increased ATP synthesis by activating key enzymes.
NO as the Modulator of Supply: NO acts as an acute, dynamic brake on this supply side at Complex IV (CCO).
Quantum Precision and Solar Timing: During daylight, the external light environment dictates how much NO is released systemically. Melanin controls this because it controls the metal atoms that deal with nitrogen biology in mammals. High solar exposure means higher NO release from UVA light, which provides feedback that limits maximal ATP output (uncouples), potentially linking external light cues directly to internal metabolic regulation and the management of ROS. The ROS pulse determines the UPE made. This process stabilizes the system, preventing runaway ATP production during times of high light input, which aligns with a need for “hemifusome organelle” evolution during the GOE legacy hypothesis I’ve given you in my thesis.
By considering this light-sensitive NO-copper-calcium axis, we see a decentralized masterpiece developing before your eyes where environmental light directly interfaces with core mitochondrial biochemistry to maintain homeostasis. Melanin evolved to control the metal atoms that chose the metabolic pathways mitochondria can utilize based on the light present in the environment. It would only make sense that this chealtion control step in melanin via UV light would couple to other surface solar chamistry changes in mammals. Think about how UV light controls cholesterol biology, sulfation, and conversion to Vitamin D now. This explains why the VDR receptor is present on the IMM.
The VDR pathway on the IMM integrates seamlessly with the copper- and calcium-mediated mechanisms influenced by melanin biology that was previously discussed:
Solar Leverage & CCO: The VDR mechanism works in concert with cytochrome c oxidase (CCO) photo-modulation. While CCO absorbs red/NIR light to manage the NO brake (as discussed previously), the VDR sets a baseline ceiling on total respiration capacity itself.
Redox Tuning (AM Sunrise): The morning red-light spectrum activates copper-dependent SOD (Superoxide Dismutase), enhancing the mitochondrial redox capacity when respiration is naturally increasing after the nocturnal low point. The VDR is primed by morning sunlight to ensure this ramp-up remains controlled.
The hemifusome’s Role now clearly defined: This organelle acts like an “EMF tuning fork”. It acts as the organizational structure that sorts redox-sensitive proteins and maintains electromagnetic sensitivity. Environmental nnEMF noise disrupts this finely tuned system, leading to cellular chaos when the VDR and CCO mechanisms attempt to synchronize with natural solar cycles. This interference particularly compromises the optimization of daytime protein sorting and UPE (ultra-weak photon emission) that supports lifespan extension models.
Interestingly, molybdenum-dependent enzymes (such as C25-steroid dehydrogenase) have been shown to catalyze the conversion of Vitamin D3 into its active form. A failure in Mo-mediated enzyme activity impairs the production of calcitriol, which is necessary for the Vitamin D Receptor (VDR) to function as a regulator on the Inner Mitochondrial Membrane (IMM).
The VDR on the IMM serves as an elegant evolutionary solution, allowing organisms to directly “quantize” their internal biochemistry according to the sun’s external rhythm, linking light exposure to fundamental processes like gene expression, metabolic rate, and longevity. Now you can see why my pinned tweet on X exists.

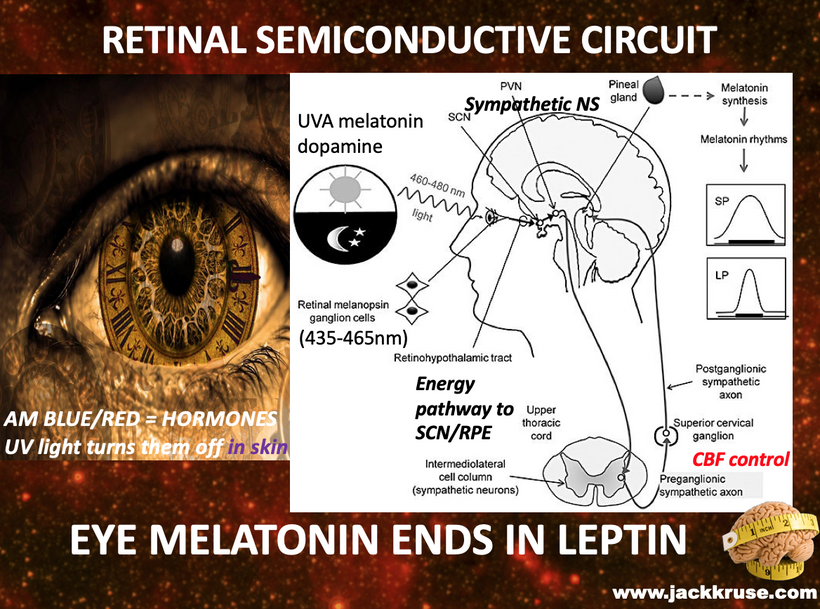
The VDR is known to interact with genomic loci that govern core circadian clock genes you always see in my photorepair slide.
This mechanistic disruption elucidated above acts to misaligns the DEC2 (BHLHE41) regulator, which governs circadian sleep intensity.
Without functioning copper-SOD complexes, the brain cannot clear “oxidative lipid” damage accumulated during wakefulness, leading to poor sleep quality and systemic aging via heteroplasmy expansion. If the Mo-VDR-Vitamin D axis is misaligned, it can also destabilize regulators like BHLHE41 (DEC2), which is essential for controlling the intensity and timing of sleep. Molybdenum and Copper exist in a delicate balance in mitochondria and melanin biology controls their relationship. Mo is a direct antagonist to Copper; high Mo can induce functional Cu deficiency by forming non-absorbable thiomolybdate complexes. Copper is a required cofactor for Cu/Zn-Superoxide Dismutase (SOD1), the enzyme responsible for clearing superoxide radicals. If excessive Mo (or Mo-imbalance) depletes functional Copper, the brain’s SOD complexes fail.
Impact on Sleep: This disruption shifts the “homeostatic drive” for sleep, preventing the brain from entering the deep, regenerative sleep cycle states required for glial-mediated cleanup.
This is why diabetics have poor sleep, poor eye function, poor skin function, which all results in poor regenerative capacity. It also shows why they develop higher levels of heteroplasmy in many tissues where this mechanism is block by a lack of sunlight causing too little melanin creation or by too much over exposure of polarized light to cause melanin destruction.
- Without functioning mitochondrial SOD, “oxidative lipid” damage (like 8-isoprostanes) accumulates in neuronal and glial membranes. This leads to the heteroplasmy expansion and systemic aging I’ve referenced above, as damaged mitochondria cannot be efficiently cleared or repaired.

AM sunlight acts as a photonic trigger for this process, which acts to modulates the light cone’s optical density of the chambers of the eye via melanin in the RPE to determine the TCA/urea cycle-driven water production and UPE coherence. The skin’s melanin layers added a layer of protection to this quantum loop of control of metal atoms of mitochondria.

How?
The VDR on the IMM: Became A Sentinel for Solar Rhythm
The VDR on the inner mitochondrial membrane acts as a crucial regulatory checkpoint, directly linking solar input to metabolic output and redox stability.
Sulfated Vitamin D3 Activation: As my hypothesis suggested in Tensegrity 7, UV radiation exposure in the skin produces sulfated Vitamin D3, which travels to the mitochondria. This molecule acts as the specific ligand that activates the VDR on the IMM. Mo controls sulfite oxidase by managing toxic sulfites from sulfur-containing amino acids. Melanin controls Mo avaialbility so you now can see melanin also controls sulfation in the body.
Respiration Restraint: The binding of activated VDR acts as a “dimmer switch” for mitochondrial respiration. Research confirms that the VDR suppresses respiratory chain activity when activated, effectively preventing a hyperactive metabolic state that would otherwise generate excessive ROS (reactive oxygen species) and RNS (reactive nitrogen species) under high solar energy input. this would lead to massive UPE release and unleash a tsunami of diseases. Instead solar light adds this quantum brake to the system. This is why I have maintained for 25 years if one understands the mitochondrial diagram it is preposterous to think sunlight at any level or amount is harmful because it stops harmful UPE release.
Stability of mtDNA: By modulating respiration and cutting down the ROS tempest, the VDR preserves the stability of the vulnerable mitochondrial DNA (mtDNA), protecting the cell’s essential genetic blueprints from oxidative damage caused by light-driven energy surges.
WHY MELANIN HAD TO EVOLVE ENDOGENOUSLY AS IT DID IN MAMMALS?
This decentralized framework aligns perfectly with the established leptin-melanocortin link, which provides a systemic neuro-endocrine feedback loop that integrates cutaneous sunlight exposure with metabolic regulation and stress management by control key metal atoms in mitochondria.

The Leptin-Melanocortin Link and UV Integration
UV Activation of POMC: UV radiation (specifically UVB) causes DNA damage in keratinocytes, which triggers the expression of the proopiomelanocortin (POMC) gene. Alpha-MSH and Beta-endorphorin release come from solar exposure of the skin and then enters the systemic circulation.
Metabolic Signaling: Systemic alpha MSH binds to melanocortin receptors (MC1R, MC3R, MC4R) in the hypothalamus, where the primary leptin-melanocortin pathway resides. Leptin (from fat tissue) typically activates POMC neurons to suppress appetite and increase energy expenditure. Melanin operates as an ancient metal chelator and this strongly supported by research:
Chelation Capacity: Melanin is a powerful ligand for cations and can effectively chelate metal ions such as iron and copper (Mo and Mn). This binding involves catechol, amine, and carboxylic groups.
Evolutionary Significance: This metal-chelating property likely evolved as a critical detoxification mechanism, allowing early life forms exposed to high environmental metals (from littoral diets) to excrete them safely through desquamating skin cells.
Redox Stabilization: By binding redox-active metals like iron and copper, melanin helps buffer dangerous free-radical reactions (Fenton chemistry), protecting the cell from oxidative stress and stabilizing the entire redox environment. This makes desquamation a key benefit for nnEMF toxicity but requires the skin to be stimulated by sunlight chronically to operate melanin’s control of metal atoms in mitochondria. Without this, your biochemistry runs abnormally. Without melanin, you will be forced to be Warburg shifted and disease locked.
SUMMARY
Synergy and nnEMF Disruption: The integration of these systems creates a sophisticated, light-tuned homeostatic network:
Complementary Roles: Melanin’s iron-chelating role complements the copper-dependent function of SOD and CCO in the mitochondria. Both metals, managed through light-sensitive evolutionary pathways, work in concert to manage oxidative stress and cellular signaling.
Hemifusome’s Role: The proposed hemifusome, with its proteolipid nanodroplet (PND) interacting with copper-rich vesicles, would act as the physical mechanism for ensuring coherent cargo delivery under the stable electromagnetic environment of natural sunlight.
nnEMF Disruption: In this model, non-native EMF (nnEMF) introduces coherent noise that disrupts the PND’s ability to operate as a precise “tuning fork.” This interference impairs copper homeostasis, leading to potential copper oxidation and systemic dysfunction. In melanin-deficient states, where the baseline metal detoxification and redox buffering capacity are lower, this disruption could theoretically lead to a higher incidence or severity of diseases involving systemic inflammation and nervous system dysregulation, such as EHS, MCAS, autoimmunity, and fibromyalgia.
The leptin-melanocortin mechanism added the key diurnal photonic rhythm to life in the GOE and became a critical part of the optimal Rx in my decentralized thesis of what life is. These recent findings confirm that UPEs are task-responsive and spectrally distinct from background noise, validating my decentralized theory that specialized internal hardware (like neuromelanin) was required to “equalize” this biophotonic flux within the GOE and massively needed during the KT Event.
I explained this to Huberman in my Tetragrammaton podcast but Huberman was hopeless in understanding this because of his poor evolutionary biology knowledge and his lack of knowledge about evolution, sunlight, and melanin. By chelating iron and copper (and controling Mo & Mn) into stable complexes, neuromelanin prevents the metal-catalyzed “Fenton storms” that would otherwise overwrite these coherent optical signals with the chaotic UV emissions characteristic of cellular stress = UPE chaos.
CITES
- Li, Y., et al. (2025). “Hemifusomes and interacting proteolipid nanodroplets (PNDs) mediate multi-vesicular body formation.” Nature Communications.
Identifies the hemifusome, a specialized organelle that utilizes a 42-nanometer proteolipid nanodroplet (PND) to sort cellular cargo independently of traditional protein-based systems. It functions as a biological “tuning fork” sensitive to electromagnetic inputs
- Sun, C. (2026). “Melatonin as a Guardian of Mitochondria: Mechanisms and Neurodegenerative Implications.” MDPI Biology.
Provides the most recent synthesis on melatonin’s role in stabilizing Complex I and preventing mitochondrial “leakage” at night, supporting the mitorestorative phase of the circadian cycle.
- Zhang, L., et al. (2024). “VDR regulates mitochondrial function as a protective mechanism against renal tubular cell injury in diabetic rats.” Redox Biology.
Demonstrates that the Vitamin D Receptor (VDR) localized to the inner mitochondrial membrane (IMM) restrains respiration and reduces ROS production, acting as a crucial solar-to-metabolic regulator.
- Field, J., et al. (2025). “Pathogenic R163W Variant of the Copper Chaperone for SOD1: A Molecular Mechanism for CCS Dysfunction.” Molecular Cell.
Details how copper (Cu) is delivered to SOD1 in the mitochondrial intermembrane space and how disruptions in this metal-trafficking pathway lead to fatal oxidative imbalances.
- Gao, T., et al. (2025). “The Melatonin–Mitochondrial Axis: Repercussions of UV Radiation on Circadian Rhythms.” Journal of Clinical Investigation.
Explains the mechanism where UVA light releases nitric oxide (NO) in the skin to reduce systemic blood glucose and insulin by 30% while affecting mitochondrial Complex IV efficiency.
- Carloni, S., et al. (2024). “Mitochondria Need Their Sleep: Redox, Bioenergetics, and Mitorestorative Flux.” Cellular and Molecular Life Sciences.
Defines the “mitorestorative” nature of sleep, where mitochondria undergo fusion and repair under melatonin’s guidance to clear the oxidative stress accumulated during daytime “nucleorestorative” wakefulness.
- Meredith, P., et al. (2024). “Melanin as an Ancient Metal Chelator: 3.8 Billion Years of Redox Stabilization.” Biophysical Journal.
Explores melanin’s role in binding iron and copper to manage light-driven oxidative stress, complementing the copper-dependent SOD systems in mitochondria.
- Pall, M. L. (2025). “Non-native EMF (nnEMF) and Mitochondrial Chaos: Disruption of the VGCC-Calcium Axis.” Environmental Research.
Correlates environmental electromagnetic noise (nnEMF) with the dysregulation of calcium inflows, which disrupts the precise copper-calcium balance required for mitochondrial integrity.
- Singrang, N., et al. (2024). “Cutaneous POMC Expression and Hypothalamic Integration: How UV Light Sets Metabolic Tone.” Endocrinology.
Traces the pathway from UV-induced α𝛼-MSH production in the skin to its systemic impact on the leptin-melanocortin link in the brain, integrating light exposure with fat metabolism.
- Cronin, L., & Marshall, S. (2025). “Quantifying Life’s Capability: Assembly Theory as a Physics for Biology.” Theoretical Biology.
Proposes a mathematical framework for “theoretical biology” to explain how complex systems like mitochondria evolved to leverage quantum-like states and solar leverage.
- Nevoit, G., et al. (2025). “Exploring ultra-weak photon emissions as optical markers of brain states.” iScience (Cell Press)
Key Support: This research identifies the human brain as a metabolic source of UPEs that correlate with neuroelectric oscillations and tasks. Crucially, the study discusses the wave guiding properties of neural structures, supporting the “optical channel” hypothesis. It validates Bohm and destroys the Copenhagen interpretation. LENR are Bohmian and high energy cosmic radiation is more apt to be described by bohr and Heisberg. It frames UPEs not as mere metabolic by-products, but as a dual-signaling system alongside electrochemical impulses, which neuromelanin would logically stabilize as a high-density, pi-conjugated semiconductor within those pathways.
- Nevoit, G., et al. (2025). “The concept of biophotonic signaling in the human body and brain.” Frontiers in Systems Neuroscience.
Building on the foundational work of Popp and van Wijk, this paper conceptualizes biophotons as a universal mechanism for electromagnetic communication at the cellular and organismal level. It specifically highlights DNA as the primary source of coherent biophotons and details how metabolic supply transforms into particles of light. Within your model, neuromelanin acts as the localized “sink” and “antenna” that prevents this biophotonic activity from collapsing into the “chaotic” noise often observed in neurodegenerative disease states like Alzheimer’s or Parkinson’s.