I have waited about ten years to unleash the ideas that are coming in this talk.
I can promise you long term members and new Patrons will be astounded by the quantum biology built into the human skin.
Last year in Vermont I decided to use the story of my wife to show the audience how obesity really begins in the eye. The talk began with the mass equivalence equation E=mc^2 and ended with a picture of her at the end of her transformation.
This was her after in May 2017 below
The paper that showed my insights to the quantum biology at work in the eye to cause obesity was publish in 2017. I had these ideas since 2003 from my work in reviewing the world wide literature.
Last year I laid the entire case out for the eye in modern disease epidemics and this year will add more meat to the bone. For a review of last years event have a review of the video on YouTube.
I won’t disappoint this year on Saturday June 2. I know where the pieces fit because I have seen them fall apart in me, my wife, my kids and thousands of patients. Now I am going to show you how to put them back together using the wisdom of nature.
Today’s mitochondriac wisdom being poured into my Vermont presentation as we speak: Truths are approximations. All of them are, because they are relative to the physics of the present moment.
Consider coming to Vermont and getting new data on how your skin really operates with your eye and mitochondria to build your optimal life. The name of the talk is “skin in the game.” My talk will focus on the poetry of nature in your skin.
Several of my members went to a bio-hacking conference in California and were told blue light exposure during the day was NOT harmful at all in 2016, and quite helpful for the human eye. This was probably the most damaging advice ever given at any event I’ve heard. Now two years later we have many PEER reviewed article pointing out just how bad manmade blue light from screens are for the human retina.
One of the things that many of the people in the conference did not put together at the time was the event organizer has a serious vested interest in selling endogenous glutathione. It turns out the more blue light you allow in your life the more you destroy endogenous glutathione cycle. So this is an example of how a wise biohacking marketer can create a market with bad advice and sell into it in a big way hoping to get bought out by even more clueless rich angels.
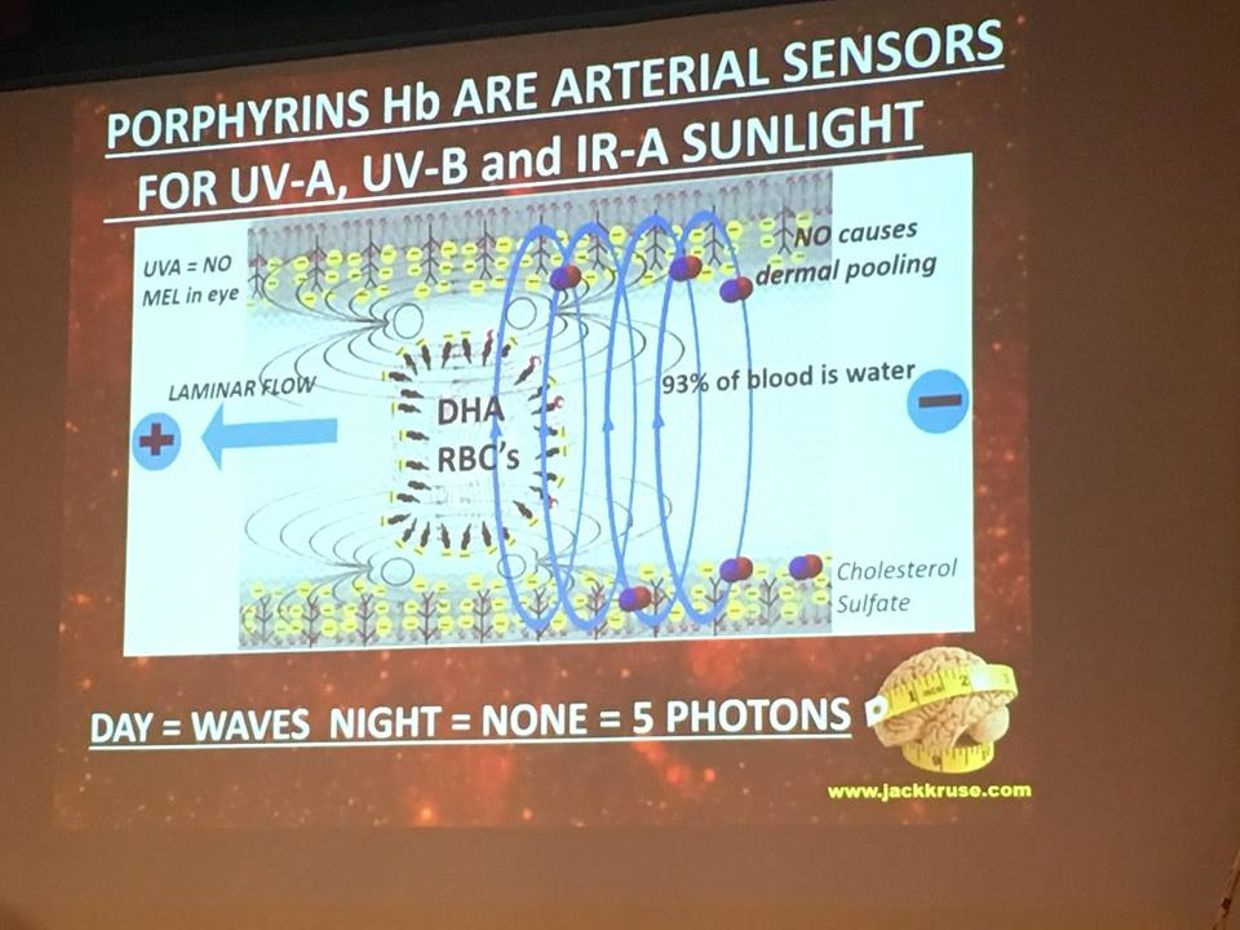
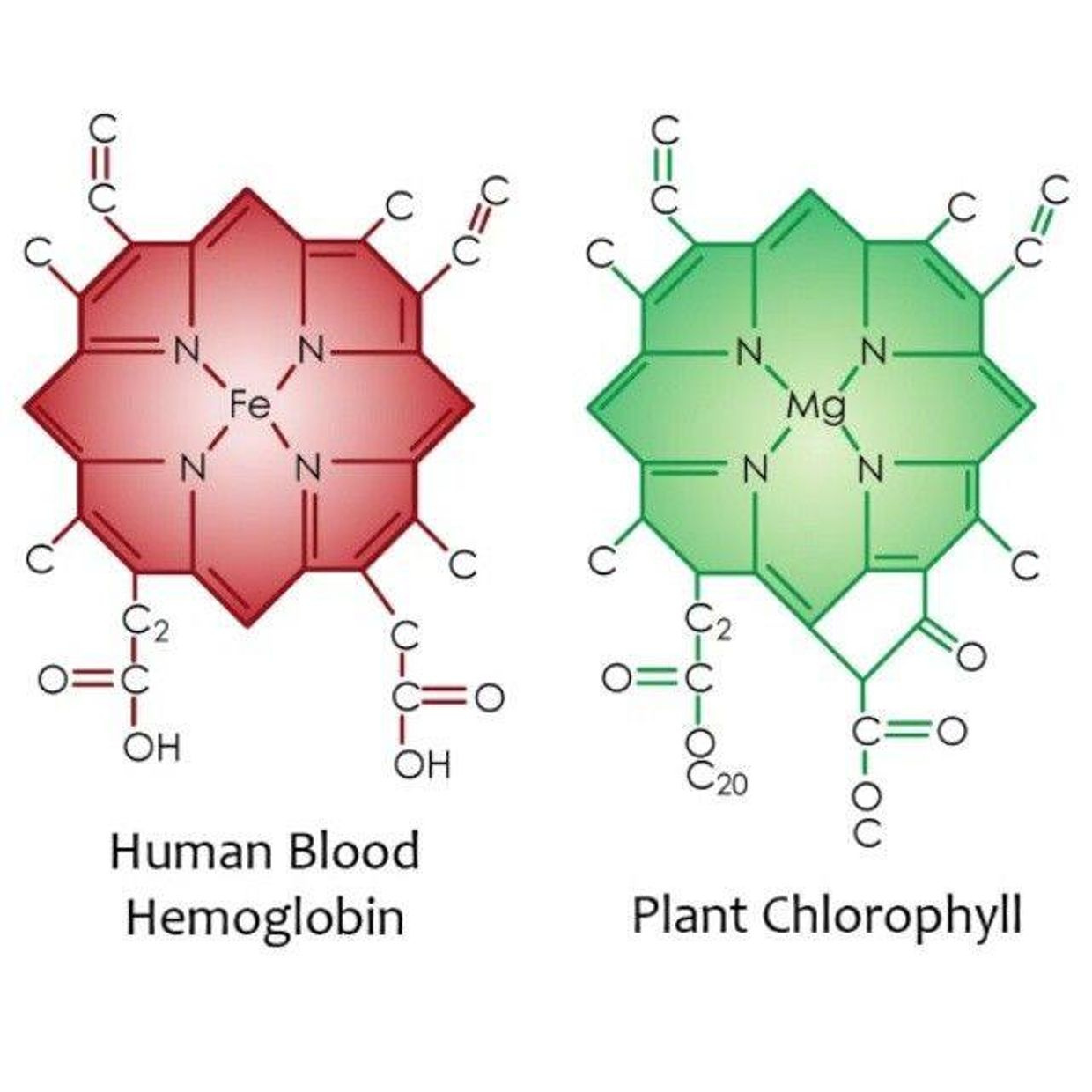
Bilirubin and glutathione have complementary antioxidant and cytoprotective roles in humans. Glutathione (GSH) is water-soluble compound made out of sulfated amino acids and primarily protects water-soluble proteins, whereas the lipophilic bilirubin protects lipids from oxidation. It turns out that when the sun shines on us the red portion of the spectrum is dominant. It makes up 42% of sunlight. Red light from the sun is the antidote to blue light damage. When red light hits our skin it makes more ATP and antioxidants like these two chemicals. Blue light creates more free radicals than any other form of light man uses in his light bulbs. There must be a balance of free radical production to keep balance of autophagy and apoptosis in a cell. Blue light destroys that balance because it lowers glutathione by destroying the sulfated amino acid cycles that work with other metabolic cycles in cells.
I’ve decided to share a few things with you over the last few years that challenges this very bad idea of using manmade blue light indoors for any reason. People have no idea that blue light can affect their baby born with jaundice in the hospital. Did you know that jaundice comes from a child born of parents who are blue light toxic? It is true. That jaundice comes from the early breakdown of the child’s porphyrins in RBC’s called hemoglobin. That process is controlled by circadian cycles. Their creation is controlled in the mitochondria. Did you know in healthcare that the light they specifically use to eliminate the yellow pigments from RBC’s breakdown is artificial blue light? Jaundice, left untreated, can cause brain damage in infants called kernicterus. I am not advocating non-treatment of jaundice, I just want you to know in the past UV light of the sun was used to reverse and protect children’s brains in hospitals. Why has the process changed, with respect to the light used?
Bad science tied to UV light in the 1950’s. So, why did they choose blue light as a replacement? The reason is simple……yellow is the color of jaundice and blue is its complementary color, therefore, phototherapy using these wavelengths. What is not well known is that blue light in children stimulates melanogenesis and hyperpigmentation that lowers the ability to handle UV light. It is even associated with more nevi and more melanoma risk longer term. This information is rather new and clearly was not presented of known at this “biohacker” event. Phototherapy induces isomerization of bilirubin rendering it extractable because it becomes water-soluble by altering the charge in the kidney’s basement membrane allowing its easy clearance via the urine and hence it is used as a routine treatment of neonatal jaundice. What the article does not tell you is that pre-1950’s full spectrum light was used and it was more effective. Then in 1959 a paper on retrolental hyperplasia of the eye showed up in the literature and caused all pediatricians to begin avoiding UV light for this reason. They linked the two incorrectly. This one paper is why today modern medicine universally thinks UV light is always toxic. It is not when it is buried with the other frequencies in sunlight.
John Ott talked about this paper in his book “Heath and Light”. You might want to read the paper and his book. Today’s literature links uveal melanoma (eye) to early blue light exposure in life. Nothing is earlier than a jaundiced baby. That child came from germ cells that were already affected by blue light before their parents even had sex.
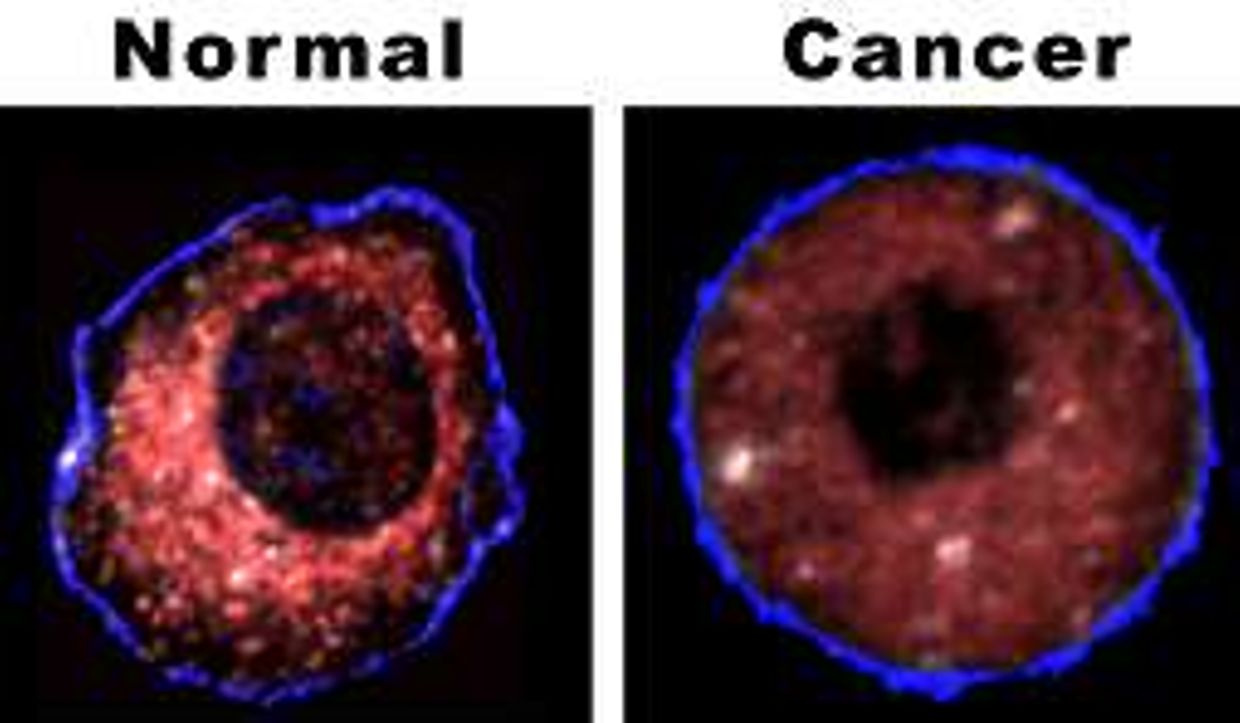
These kids, when they are born today, are all baked under blue light to avoid kernicterus. Ultraviolet radiation does not figure prominently among the risk factors for ocular melanoma, but blue light IS deeply linked to this new fast-growing cancer of the eye. Eye melanoma is the fastest growing cancer of the eye today. Guess why? The picture below contains the answer.
Blue light is behind its cause. My latest Quantum Thermodynamics #9 blog explains to you why it is happening all around you today.
If you look at the above picture you might understand now why we are seeing jaundiced babies grow up to face precocious puberty with early menarche in girls with early fertility damage in their germ lines, poor sperm development, boys become more like girls, and girls developing bad cramping during periods with heavy bleeding as they grow into women. Both wind up needing glasses for myopia before too long too. As they age they get proliferative diseases of their eyes and skin as a long term result. No one sees where the pieces fit until they understand how they fall apart.
This interesting observation of blue light effects to ocular melanocytes was followed up by a study that sought to mimic the effect of blue light on UM cells within the context of the mammalian eye. Human UM cells were xenografted into the eye of an albino rabbit model of ocular melanoma and subsequently exposed to blue light showed enhanced proliferation upon removal and recapture, compared with control samples protected from blue light. The significance of this finding is that the UM cells were exposed to blue light while residing within the choroid, effectively demonstrating that blue light affects uveal cells and can enhance their mitotic ability. This is a crucial step in linking blue light to malignant changes within uveal melanocytes in vivo. The final confirmation of the link between blue light and UM in vivo came from a study in Long Evans rats, a strain with pigmented eyes in which there have been no reported cases of intraocular melanoma. This study described the development of an ocular tumor in one animal following blue light exposure (434–475 nm). This is the range of the melanopsin receptor in the eye known to control melatonin production in the eye to control the entire central retinal pathways to the SCN. You’ll be hearing a lot about that receptor next week in Vermont.
Are the LCHF experts correct about biochemistry and cancer with respect to food dogma?
NOPE
NON PATHOLOGICWARBURG SHIFT = cancer cells tend to convert glucose to lactate despite the presence of oxygen. Warburg called this phenomenon ‘aerobic glycolysis’, a term that is now synonymous with the ‘Warburg effect.’
The ‘Pasteur effect’, is where oxygen inhibits glycolysis, or conversely, hypoxia or a pseudohypoxia stimulates glycolysis. This last effect is why diabetes and higher Hba1C’s are seen with nnEMF of all types. Why? All nnEMF stimulate and cause pseudohypoxia in all cells lines but the effect is different in cell lines. High HbAIc’s are very dangerous to tissues with high metabolic rates that use the TCA cycle for beta-oxidation.
So are there any cells that are obligatory anaerobic glycolytic cells that are not cancerous in healthy people? Yes, there are and they all have one thing in common. Theses tissues exist within a life with low oxygen demand and therefore they do not make a lot of ROS/RNS free radicals to operate. It seems they do not need many electrons from ECT transport but they seem to crave protons. On the surface this seems like an unusual set of circumstances but it is not. Some of these cells do not have mitochondria and the ones that do hardly ever use the TCA or urea cycle. Obligatory glycolytic cells in normal tissues exist with autophagy and apoptotic intact as growth controls.
When we lose control of autophagy and apoptosis cells become at risk for having to use a PATHOLOGIC Warburg metabolism that has been the calling card for oncogenesis.
All somatic cells in humans are committed to self-sacrifice to protect the germ line. This is why apoptosis evolved initially. The key is not all cells share the same risk in this game of life. Some cells lack mitochondria or good blood flow, and as a result, they need glucose to run basic energy programs to remain biotic. This shows you that life is not wholly dependent on a lot of energy flux as we all believe. Is there something else that cells who have growth control who need glucose can live off of? There is. Remember these obligatory glycolytic cells don’t stop working during starvation either because they use gluconeogenesis for biosynthesis. What feeds them the information to know how to operate in this way? The information from solar light via the electron and proton spin, is the answer. These are the cells in the body that use information quanta more than they need energy quanta to survive.
What cells are these? Red blood cells, renal medullary cells, and certain cells in the retina called Muller cells and the cones used for color vision.
It gets more interesting. Warburg himself, and his team, also noted that normal mammalian retinal explants displayed obligate aerobic glycolysis normally. In other words, the retina, under normal solar conditions exhibits a metabolism that is similar to cancer cells. This was shocking to me when I first heard it 25 years ago. Why is that?
Why would our eyes and cancer cells share a metabolism?
This retinal finding, however, did not fit neatly with Warburg’s beliefs about cancer pathogenesis. In fact in the 1920’s Warburg’s critics used this against him and his thesis at every turn. Most of the critics and skeptics convinced him and his team that it HAD TO be attributed to an experimental artifact. Was it really caused by this, or was it related to the fact that all these obligate NORMAL glycolytic cells have to work this way for a reason? Was the reason due to the retina being heavily involved in light transduction metabolism? Might it be any cell that is using obligate glycolysis does so as a safety fuse because they are doing something unusual with incident light waves? Several decades thereafter, researchers with better tools confirmed that the mammalian retina indeed displays a strong NON-pathologic Warburg effect. the non pathologic Warburg shift does not allow for serious growth or proliferation. It can keep growth and metabolism under control. This is why you should never pay attention to critics/skeptics. Warburg’s observations were correct. He was never able to explain them and today I am going too in this blog using quantum biology of mitochondrial function.
REALLY?
HOW?
Might we be able to use this unusual finding to learn more about the biophysics and the quantum biology of human beings? I think the answer is unquestionably yes. Cancer researchers today routinely report that the Warburg metabolism is unique to cancer. Nothing could be further from the truth when you examine the data of the human retina. Today we know the Warburg effect is widely described in other cell types, namely embryonic stem cells, human T lymphocytes, neutrophils, dendritic cells and macrophages. This should begin to peak your interest after my April 2018 webinar. Are all these cell types deuterium bombs? Ye,s they are for the most part.
All life on Earth is believed to use adenosine triphosphate (ATP) to transfer energy. After the April 2018 webinar you now know definitively, I do not hold this view point. Let me give you a quick review. ATP is generated via two related metabolic pathways: OX-PHOS and glycolysis. Glycolysis converts a single molecule of glucose into two molecules of pyruvate, generating two ATP molecules. The final step requires pyruvate kinase (PK), which exists as several isoforms, notably PKM1 and PKM2 in the cytosol. In the presence of oxygen, pyruvate is usually converted to acetyl CoA, which then enters the Krebs cycle in the matrix, forming electron donors for OX-PHOS, generating approximately 32 net ATP molecules. When oxygen is scarce (pseudohypoxia) or falls short of demand, ECT is slowed. When ECT is slowed or absent, autophagy becomes impossible or unlikely, and pyruvate is shunted away from OX-PHOS in the TCA cycle and is converted into lactate by lactate dehydrogenase (LDH) in the cytosol to regenerate nicotinamide adenine dinucleotide (NAD+). Remember NAD+ wants all its H in the H+ form from the matrix. It does this because it needs the information of the light photon to transfer to the orbital angular momentum of the proton. This is the electron carrier of cytochrome one that deliever electrons to oxygen. Each electron carries photon energy and information as you learned in April 2018 webinar.
Each of the steps within the glycolytic pathway is catalyzed by a specific enzyme or enzyme complex. This is not important right now. Some of these enzymes have a role in protein transcription regulation, cell motility and apoptosis regulation. Cytochrome c is the controlling mechanism for human apoptosis. If apoptosis is operational it is impossible to get cancer. Apoptosis is made efficient by IR and UVa light. This is important and critical for the mitochondriac to know and understand how it fits into the story of quantum biology using quantum thermodynamics. Information to these enzymes is far more important than energy flux. It seems the process of apoptosis needs more information to operate than energy flux.
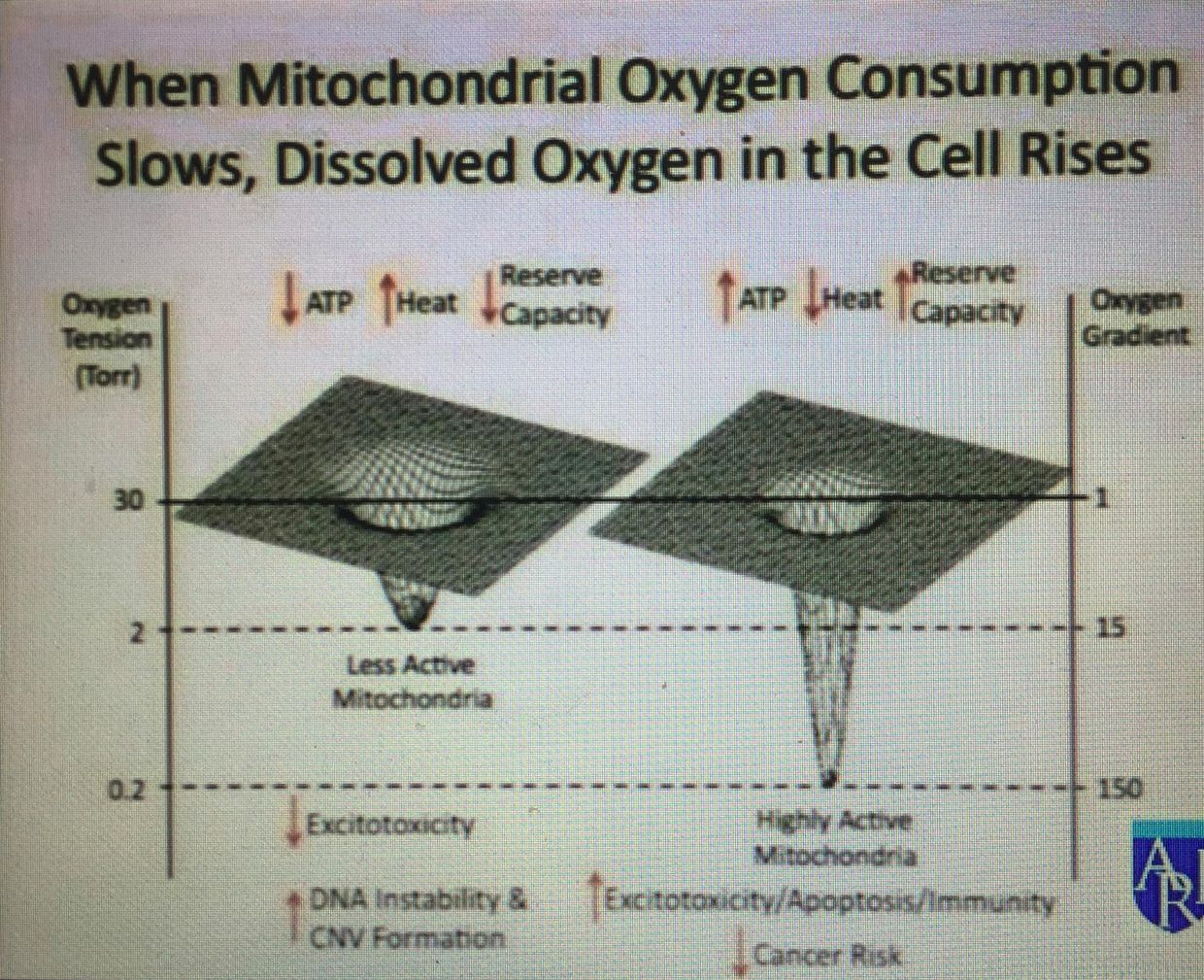
I wrote the EMF 4 blog post long ago to teach you about the pentose phosphate pathway (PPP). I did this for many reasons. I was trying to get you to realize that a cell has many trap doors, or quantum demons, it can use when the environment changes light and temperature and calls for them to be used. Anytime light and temperture what also changes in the environment? Oxygen tensions do. Mitochondria pay DEEP attention to that change as a picture below shows. These are the metabolic doors that appear to vanish when they are not needed. When they are needed they can be massively amplified, as you saw in the May 2018 webinar. When a cell is missing IRA and UVA light is can cause a massive amplification of things that a cell should rarely see and this is why cancer is often the result of a loss of control of information quanta. The reason for the observed biochemical changes are always biophysical and way below a researchers ability to find them because they have no idea they are there, much less what they do in cells. You cannot find or control for things you do not even know that matter.
In proliferating cells, glucose not only produces ATP, but also provides metabolic intermediates for biosynthesis. Intracellular glucose can also be directed towards biosynthesis: into the pentose phosphate pathway to generate nucleotides and nicotinamide adenine dinucleotide phosphate (NADP), or gluciose can be reduced, to make the amino acids, serine and glycine. This was the serine glycine interconversion pathway I mentioned in the Reality series. Serine and glycine can link and jump from glycolysis at the phosphoglycerate step. The enzyme phosphoserine phosphatase is the final step in glucose-serine conversion. The PPP has to use NADPH. The H has to be H+ because it is the key information quibit. It carries more information from the environment to the matrix of a cell than dueterium can. This is exactly what I said above about NADH. This detail is critical to understand.
The ability to oscillate between biosynthesis, energy production, and information transfer is quite unique in cell biology. It provides proliferating tissues with a powerful metabolic strategy known as the ‘metabolic budget system.’ This is kind of like a carburator controlling the flow of gas into an engine cyclinder where a spark plug is ready to produce power. This phenomenon actually goes hand in hand with what Warburg described in his papers on these cell effects. This strategy can be viewed as the presence of the Warburg effect in a tissue using glucose for biosynthesis. Such a phenomenon, is rarely noted in a non-proliferating tissue, such as the retina or RBC. These two tissues were my focus in the last two years in the my Vermont talks.
The glycine serine pathways (talked about in survivor soup blog) are needed in making all the opsins proteins in the body. Few people seem to know this fact either. Pyruvate kinase isoform M2 (PKM2) and hypoxia- inducible factor-1 (HIF-1) are the key regulators of the non pathologic and the pathologic Warburg effects. HIF-1 is a protein that stabilizes 3D protein conformation. The control of oxygen is under the control of local tissue metobolism. Methionine cycling is critical in the angiogenesis signal in tissues. As the TCA and urea cycle slow, methionine is raised as a collateral effect. This is incredibly important in the opsin systems because of the potential of photooxidation from the highly powered light like the blue light at night. HIF is induced in hypoxic or pseudohypoxic environments.
Pyruvate kinase (PK) is a glycolytic enzyme that catalyses the conversion of phosphoenolpyruvate into pyruvate, generating one molecule of ATP in the rate-limiting final step of glycolysis. There are four isoforms of PK in mammals: L – liver, R – red blood cell, M1 – adult (muscle and brain) and M2 – embryonic and tumor. Cells needs multiple isoforms to control the metabolic rate of each tissue differently to modulate growth. How do they do this? Each one allows a different level of deuterium to get into the Kreb’s bicycle area of each tissue.
GEEK PART: Here is where it gets interesting and complex for the non scientist. Uniquely, PKM2 has an allosteric pocket not present in the other isoforms that permits binding to phosphotyrosine peptides and fructose 1,6 biphosphate. This structural configuration renders PKM2 vulnerable to various regulatory inputs. Whereas PKM1 forms a stable, constitutively active tetramer, PKM2 oscillates between the active tetrameric and the inactive dimeric (or monomeric) forms. The dimeric form has a low affinity for the substrate phosphoenolpyruvate and lower activity than the tetrameric form. When the dimeric form dominates, phosphoenolpyruvate conversion becomes inefficient, and as a consequence glycolytic intermediates upstream of phosphoenolpyruvate accumulate and are available for biomass synthesis and cell proliferation. This is why cancer has to depend on glycolytic intermediates………If the TCA/urea cycle is non operational this pathways to biosynthesis and survival is its only choice unless the cell can recpature control of autophagy or apoptosis in some way quickly.
As the glycolytic intermediate, fructose 1,6 biphosphate accumulates, the reaction favors conversion of the dimeric form back to the tetrameric form and pyruvate is produced efficiently again. These regulations of PKM2, labelled as the ‘metabolic budget system’, has been proposed to control the anabolic biosynthesis versus energy production in tumor metabolism.
Guess what I believe controls this process? The information in protons and electrons that have had photonic information transferred from the quantum spin number to the orbital angular momentum (OAM). Oxygen tension also play a role as the picture below shows. This is how intelligence is built in atoms in these cells. Apoptosis especially uses this mechanism, as I laid out in the May 2018 webinar.
High PK activity leads to depletion of glycolytic intermediates available for biosynthesis and therefore impairs cellular proliferation and growth in obligate glycolytic cells like the RBC’s in the blood. This is why so many cancers are associated with anemia. Ironically, the tumor tries to get around the anemia by increasing angiogenesis factors released from the surface of arteries by rising levels of anion amino acids with sulfur. As anemia worsens the chance of apoptosis working well drop signficantly because RBC’s are what carry IRA and UVA light to restore apoptosis efficiency to the mitochondria in our tissues.
The situation I am laying out here would not be a good state for a cancer cell. There is a lesson here in cancer prevention. Cancers by default, need a ton of glycolytic intermedates to make things it needs to divide, so it always wants high activity of PK. To keep that chronic growth activity in cancer, autophagy has to be impaired and apoptosis has to be eliminated completely while ECT flow has to be brisk. How does cancer accomplish this? You cells have to bring more oxygen to the cancer cell and the excess oxygen pulls electrons briskly to oxygen because of its electronegativity. This augments ECT even in a broken mitochondria. What causes this amplification of oxygen/angiogenesis when a cell is using glycolysis and the PPP? Methionine cycle kinetics do. Methionine is an essential amino acids loaded with sulfur. It turns out when the TCA and urea cycle kinetics are broken, methionine levels rise and becomes a relative toxin because as it rises in a tissue it cause angiogenesis. Cells forced to use the two older metabolic pathways should never have high oxygen tensions because this always leads to a proliferative pro-growth signal from the mitochondria to the nuclear genome.
It also explains why glucose is upregulated in cancer cells or in obligate glycolytic cells. It is not because cancer cells only use glucose and glutamine, it is because they cannot use the TCA and urea cycle because their kinetics are blocked for some reason. Those two cycles need both autophagy or apoptosis to have some minimal efficency to protect from uncontrolled growth and control of methionine. The information of how to use these metabolic pathways in this way is buried in the type of protons used in the pathways. Hydrogen isoforms is the optical switch in these utilization of any metabolic pathway in a cell. When the signal is disordered cells begin to use older evolutionary pathways when oxygen tension is too high. When this occurs, deuterium leaches from cell membranes into the matrix where Kreb’s bicycle sits, and it destroys TCA and urea cycle kinetics faster than anything on Earth. The amount of deuterium let in by a cell via uncoupling proteins and oxygen levels determines what metabolic path will be chosen in an environment: It also determines what pathway is optimal for a non proliferation or proliferation tissue.
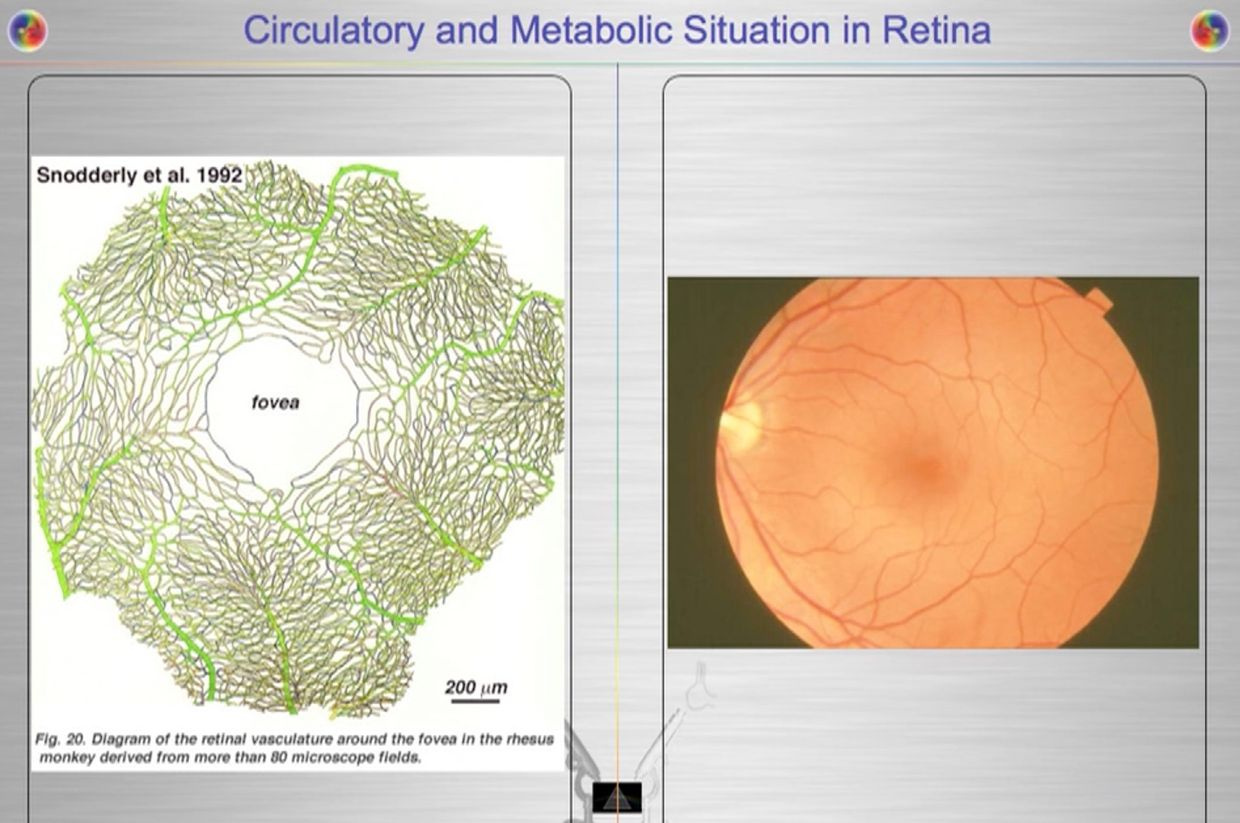
In the RBC’s and retina evolution controlled the choice by making mitochondria and blood flow scarce. Given the situation, we certainly do not want light from the sun causing uncontrolled growth in our eye, because then vision is obscured. But this i sexactly what happens in a diabetic retina. To avoid this situation, the body uses the control of proton flow in the matrix and ATPase to control these pathways in a non proliferative way. Not all the cells in the retina use the TCA level because of this. Muller cells happen to be one of these cells (pic above).
This is why RBC’s and parts of the retina have no mitochondria and little blood flow in the fovea where sharp color vision occurs. It is shocking to see such a high energy demand tissue rely on older metabolic pathways to limit oxygen to control growth. Could this be why people with faulty mitochondria develop sleep apnea too? Is it a protective phenomena built by nature? I think so.
The retina has prodigious energy demands to maintain the neurons in an excitable state for phototransduction and neurotransmission, in addition to the maintenance of normal cellular function. Any cells involved in light transfer seems to have a high concentration of lactate in the venous blood while also having low oxygen tensions. This is a reason I use lactated Ringer’s solutions when I am doing mitohacks involving light frequecies when my BUN/creatine ratios are altered. These labs are redox proxy’s telling the mitohacker that there is hypoxia in a cell and we better be careful in how we construct our experiment. I had to leanr this lesson the hard way many times in hacks. IV LR solution is an awesome hack for people with chronic BUN/creatine decline. You can learn a lot about proton information capapbilities when you use an electroretinogram to educate your mitochondriac perspective (pic below).
Your opthalmic artery feeds the retina a lot more than energy and light to the eye and brain. The retina is built backwards to slow light down to harvest a massive amount of its information to build things and to know things about the environment we inhabit. It can be seen on direct examination of the eye. Recall that 60% of your circulating blood volume passes through this artery in sunlight. This is a massive way to add energy and information to the system of the eye and brain. The blueprint of retinal physiology is telling me that the retina is more interested in information in light waves than its energy because the total amount of oxygen extracted from choroidal (RPE) and retinal blood flow combined has only accounted for partial accounting of the oxidation of glucose in hacks I have done in this tissue. I found out that the same experiments have been done in many mammal retina’s because of Warburg’s original papers were where the obligate glycolysis was first observed in science. In pigs complete oxidation of 37% of all the extracted glucose, were found in their retina and this reflects the high glycolytic activities in the pig retina when light was present. In pigs, the majority of the glycolytic substrate was derived from the choroidal circulation (RPE), indicating there was greater metabolic activity in the outer retina where vision DID not happen. This is where melanopsin was located. This was a big clue to me why blue light is devasting to growth and metabolism choices in an animal. It also explained why cancer was a serious consequence of blue light at night.
When I learned about these counterintuitive concepts I knew immediately the reason why it localized to the outer part of the retina. This is where melanopsin is plentiful. All opsins are connected to a Vitamin A moiety. Diurnal mammals and nocturanl mammals have this but the covalent binding of Vitamin A to the melanopsin is radically different. I began to realize why. That bond was stronger to offset the risks of light at night when you are nocturnal to control growth. This opsin needed Warburg protection to operate safely in the mammalian retina. It made perfect sense when I thought about the consequences if the systems pieces fell apart. Blue light is highly powered and will make a lot of ROS and not much ATP. Lowered ATP means lowered antioxidant spikes to cause swelling, which is the stimulus to growth. This mechanism had to have tight controls otherwise cancer is very likely in the animal.
Excessive ROS production is dangerous to the retina because it will stimulate apoptosis and thin the retina . So what did Mother Nature do to protect us?
As I laid out in my Vermont 2017 talk, she put the photoreceptor far away from the blood supply. This put a ton of blood, water, and deuterium between the two positions. This allowed massive information transfer from sunlight to the H+ protons while the heavier atomic mass of deuterium moves waves of blood through tiny vessels more quickly using solitons. The ROS of the blue light was quenched by the presence of 42% red light and because the cells in this region of the retina had to be obligate glycolytic cells that would make low levels or ROS to protect from an apoptotic signal. Mother nature is nothing short of amazing. All of these quantum thermodynamic actions formed the basis of my Vermont 2017 talk.
You might want to go back and relisten to the Vermont 2017 video now that you have this understanding now.
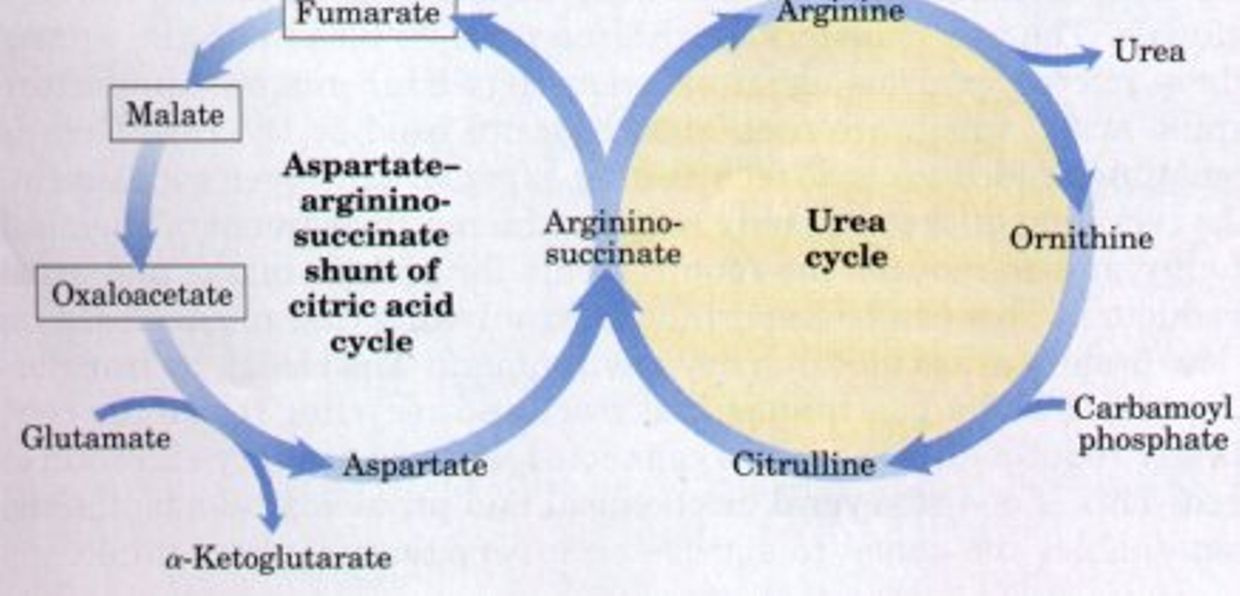
The “Krebs bicycle,” composed of the urea cycle on the right, which meshes with the aspartate-argininosuccinate shunt of the citric acid cycle on the left. Fumarate produced in the cytosol by argininosuccinate lyase of the urea cycle enters the citric acid cycle in the mitochondrion and is converted in several steps to oxaloacetate. Oxaloacetate accepts an amino group from glutamate by transamination, and the aspartate thus formed leaves the mitochondrion and donates its amino group to the urea cycle in the argininosuccinate synthetase reaction. Intermediates in the citric acid cycle are boxed.
The oxidation of acetyl-CoA to CO2by the TCA1 cycle is the central process in energy metabolism. However, the TCA cycle also functions in biosynthetic pathways in which intermediates leave the cycle to be converted primarily to glucose, fatty acids, or non-essential amino acids. If TCA cycle anions are removed from the cycle they must be replaced to permit its continued function. This process is termed anaplerosis.
Because the TCA cycle cannot fully oxidize 4- and 5-carbon compounds, these intermediates must be removed from the cycle by a process termed cataplerosis. Cataplerosis may be linked to biosynthetic processes such as gluconeogenesis in the liver and kidney cortex, fatty acid synthesis in the liver, and glyceroneogenesis in adipose tissue.
Cataplerotic enzymes present in many mammalian tissues include P-enolpyruvate carboxykinase (PEPCK), glutamate dehydrogenase, aspartate aminotransferase, and citrate lyase.
Instead of giving you a boring biochemistry lesson I am going to use a hormone that most of you know to help explain how the anaplerosis/cataplerosis cycles in the TCA cycle affect the urea cycle and can later the methionine cycles to help explain many things about life you might not have realized are going on. You really do not need to understand the steps you just need to understand the overal effects to understand your life.
Let me show you how testosterone, cell growth, all link to the sun cycle diurnally via the mitochondria to explain the TCA/urea cycle in a larger context.
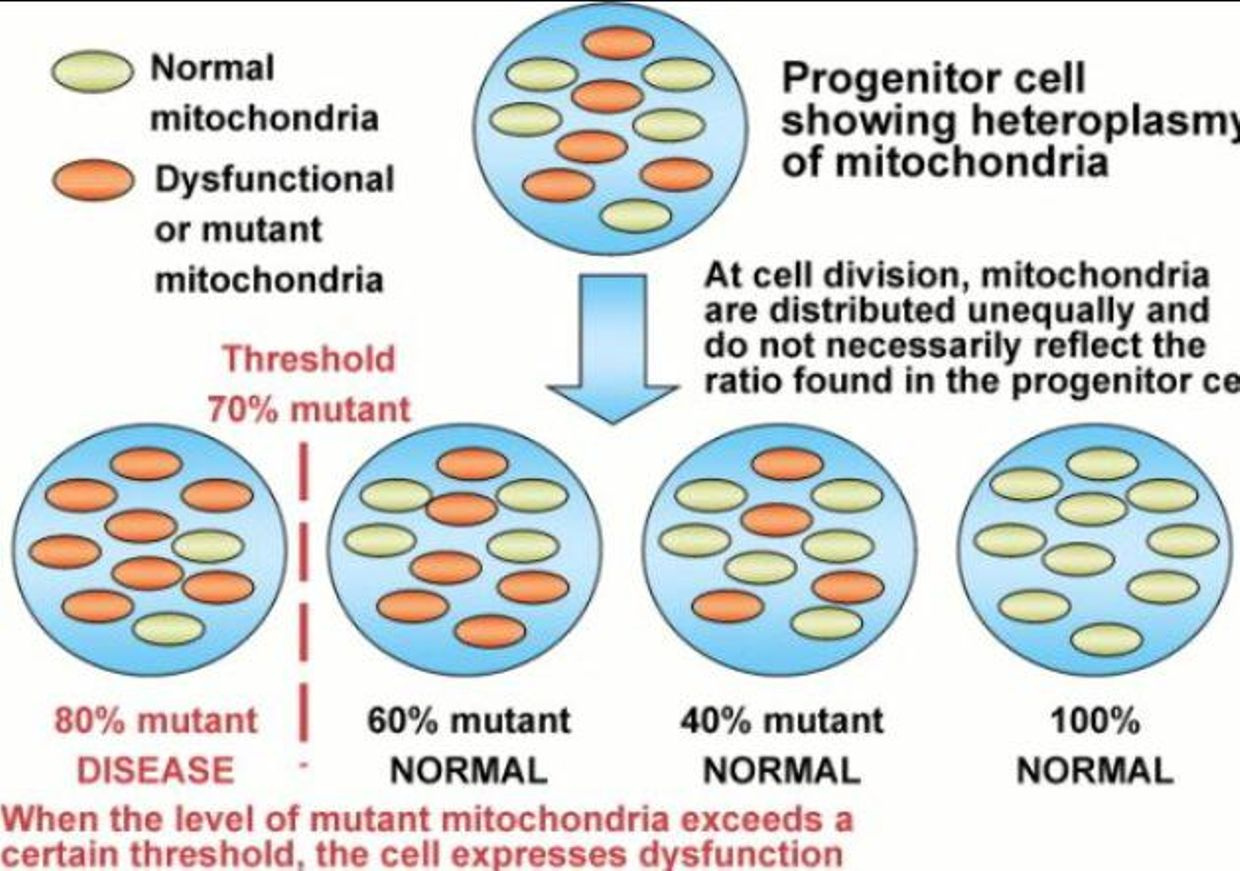
As we age does testosterone drops in both sexes to protect us from mitochondrial diseases which spike as we age and heteroplasmy rises. This rise in heteroplasmy is a maker for poor autophagy and apoptosis in mitochondria. While it’s good to have a decent immune response to pathogens, an overreaction to them — as occurs in highly virulent influenza strains, SARS, dengue and many other diseases — the repsonse to pathogens can be more damaging than the pathogen itself depending upon the state of your inner mitochondrial membrane. This shocks many people when they hear it for the first time.
This actually occured in the 1918 flu epidemic in humans. Those with the strongest immune systems tended to die more rapidly and more consistently (men). Women, children and the elderly had much better survival than young men. Why did this happen? In 1918 most young healthy men were working inside of factories under new electric lights and not in the sun.
What people have forgotten is that chronic exposure of the skin to UV light tends to limit the production of sex steroid hormones in our eyes and skin by way of our blood plasma. So men with high solar exposure should be expected to have lower sex steroid hormones. Sex steroid hormones are progrowth and facilitate biosynthesis from the TCA and urea cycles if Vitamin D levels are low. If Vitamin D levels are high it puts a cap on biosynthesis and growth via the cell cycle. It turns our Vitamin D3 and the VDR receptor curb cell growth directly on the inner mitochondrial membrane via two processes call anaplerosis and cataplerosis.
We also know that we need more sunlight as we age and we know that all cause mortality is reduced by sunlight. This hints to us that exogenous testosterone might be something we need to be careful with as we age or as our Vitamin D levels drop for any reason. I do not believe exogenous testosterone use is always bad to use as one ages but it must be put in context of the persons haplotype, SNP/SAP’s and their current environmental choices. Sunlight exposure of the skin is one of the major factors I look at.
Women have to pass epigenetic information to children which men do not have to do so, a woman’s immune system is built to be more sensitive and specific to program their offspring. This is also why women tend to get more autoimmune conditions as well. When they are in environments that cause immune activation from electropollution we should expect more Autoimmune conditions to manifest in them when Vitamin D3 levels drop. This facilitates a progrowth bias in their immune systems with poor controls.
We should also realize because of these relationships that solar exposure of the skin and eye are nature’s best vaccine for prevention of these conditions but few people really understand how the pieces fit in humans because they do not understand light of the sun well on the inner mitochondrial membrane.
In fact, the rapid, nongenomic effects of vitamin D3 appear to be mediated by the VDR directly on the inner mitochondrial membrane. The key activity of the Vitamin D receptor is to inhibit ECT to slow electron flow on this membrane. This is the breaking mechanism fine tunes apoptosis efficiency to keep a cap on uncontrolled growth in tissues with high oxygen tensions who use the TCA cycle for biosynthesis. As the VDR receptor lowers ECT flow, the distal ATPAse can be augmented by the 42% of red light in the sun to still affect the spin of the ATPase Fo head and affect the 4 red light chromophores on cytochrome c oxidase.
The inner mitochondrial membrane contains many copies of a protein called the F0-F1 ATPase. This is also called ATP synthase. It consists of two parts: the Fo component spans the membrane and provides a channel for protons to move into the matrix from the intermembrane space. The F1 component is a complex of five proteins with the composition α3β3γδε, with a molecular weight of about 360,000. This remarkable complex couples movement of H+ to the synthesis of ATP.
The ECT pumps H+ ions out of the matrix into the intermembrane space. Because the H+ ions move without counter ions, this movement is an outward current that separates charge, and therefore there is a potential developed across the inner mitochondrial membrane. This potential varies depending on the state of mitochondrial activity, but a typical value is about 160 mV, negative inside. In addition to the potential, there is a concentration difference in H+ established across the membrane. The pH of the intermembrane space is about 7.0, whereas the pH of the matrix is about 8.0. The pH differences also has huge implications on favoring certain isoforms of hydrogen. Consider the following example to improve your understanding of these nuances.
The pH at room temperature is ~7 meaning there are about 10^-7 moles of H+ per liter of water at room temperature. As the temperature increases, the ability of water to ionize in this way INCREASES and so the concentration of H+ in an aqueous solution in a cell will ALSO increase and hence the pH will drop.
The physical chemistry here all favors the formation of H+ bonds over deuterium bonds in an aqueous heat bath we call a cell. This is called the chiral effect. You’ll be getting a full blog on that topic soon enough.
Just how big a deal is it when you are a true blue mitochondriac? In all cases, raising the temperature, invokes thermal vibrational and entropic effects in all proteins, lipids, and carbohydrates in a cell. This tends to preferentially stabilize H+ over D bonds in mitochondria. This implies that heating favors H+. This is why mammals release heat from the mitochondria. Any place a mitochondria is located in a mammal they want to keep deuterium concentrations low. This chiral heating effect keeps deuterium free to roam in the ECF and blood plasma so it can perform its duties not confined to the boudaries of the kinetic isotope effect. What cells dominates the blood plasma? An RBC. RBC’s have no mitochondria.
The link between the VDR receptor and NO production is very strong in the skin and the eye. Many steroid receptors and nuclear transcription factors enter the mitochondrial compartment, where they either exert transcriptional regulation of mitochondrial DNA or control mitochondrial biogenesis and metabolism. These facts show why I staunchly believe that sunlight is nature’s vaccine for most mitochondrial diseases. Based on my reading of the literature and observations in hacks, I have concluded that the VDR is highly protective against mitochondrial disease from high heteroplasmy rates. THe VDR needs high levels of sulfated Vitamin D3 to operate properly.
When Vitamin D3 and the VDR bind some amazing things occur inside of your mitochondria.
Mitochondria need less electrons from food substrates because the function of Vitamin D3 bonding to the VDR restrains mitochondrial respiratory activity, and this lowers ROS and RNS. This occurs independent of any other factor in mammals.
But the key feature is that the VDR receptor is the main regulator or carburator that allows the cell to spare metabolic intermediates from the TCA and urea cycle. Anaplerosis is the act of replenishing TCA cycle intermediates and cataplerosis is the term used for removal of TCA intermediates for biosynthesis. The VDR receptor inhibits cataplerosis and this should make sense to you now.As you remove substrate from the main beta oxidative pathways you need to control the flux of electron flow in the inner mitochondrial membrane to put a cap on ROS/RNS and oxygen needs.
Cataplerosis removes intermediates for biosynthesis in normal growth to which may be diverted from oxidative metabolism toward a biosynthetic fate, supporting cell growth. This process has no brake in cancer because the VDR receptor only operates with a base level of sulfated Vitamin D3. Without it, the VDR receptor is inactivated and this single factor alone supports high ECT, with inactivation of apoptosis. It also allows the chronic removal of TCA and urea cycle intermediate to support uncontrolled growth. This is what we see in cancers, autoimmune conditions, and obesity.
Cataplerotic reactions effectively allow your cells to steal the 4-carbon intermediates from the TCA for biosynthesis to support growth.
REMEMBER RBC’s HAVE NO MITOCHONDRIA. WHAT ELSE MAKES THEM SPECIAL?
RBC’s use a Warburg metabolism. Yes, you heard that correctly. Most people associate that metabolism with cancer but it comes in two versions. Pathologic and non pathologic.
Cells with non pathologic Warburg metabolisms use gluconeogenesis. Gluconeogenesis is an overall cataplerotic reaction because it steals oxaloacetate to eventually form glucose. RBC’s, Muller cells, oligodendroglia cells, and embryonic stem cells use gluconeogenesis. Oligodendroglia cells make myelin in our brain. The non pathologic version occurs when oxygen levels are lowered and controlled this and it limits net cell growth.
When oxygen is raised in these cells this supports strong cell growth that can be uncontrolled. In the case of stem cells, you need to keep oxygen levels low to keep them quiescent until we need them. This means ESC cannot and should not use TCA/urea cycles to exist. In fact. ESC have immature mitochondria that cannot use the TCA or urea cycle by design. They have to rely on the older evolutionary pathways of glycolysis and the PPP to operate when the body needs them to jump into action.
WHAT HAPPENS IN THE TCA CYCLE DURING BIOSYNTHESIS?
Aspartate biosynthesis also requires oxaloacetate, so when aspartate is synthesized, the result is less net oxaloacetate. When cataplerotic reactions occur (in terms of TCA), there is less oxaloacetate to condense with acetyl CoA. This is why proliferative retinal disorders show up in diabetics with obesity and low Vitamin D levels. It is also why AI’s spike with low D3 levels. It is also why brain tumors and breast cancer spike with low vitamin D levels. Sunlight is the brake for cataplerosis in cells who use the TCA or urea cycles. That brake requires a MINIMAL level of Vitamin D3 production to control unfettered growth to bond with the VDR to slow ECT flow and increase apoptosis. Now think about what I told you in the May 2018 webinar carefully.
Women, with their more robust immune responses, are twice as susceptible as men to death from the systemic inflammatory overdrive called sepsis. This is something to keep your eyes on as 5G takes over because we should see more women die from sepsis in big cities if these links to the immune system hold true. Men are designed to have a slightly weakened immune system than women to balance growth and cataplerosis carefully. A slightly weakened immune system seems to favor anaplerosis over cataplerosis in the TCA cycle. This curbs unfettered growth assuming the VDR receptor is activated. This might be why men have more body hair than women and it is likely why women have less on their skin. Hair on the genital region would also lower solar exposure to support growth of the germline in both sexes via the Viamin D and VDR mechanism. Strong sunlight tends to lower fertility. Strong blue light toxicity does the same.
So perhaps having a somewhat weakened (but not too weak) immune system can prove more lifesaving than life-threatening for a dominant male in the prime of life. UV light increases your immune system, and testosterone seems to lower it. I believe it is designed to be dynamic to react to the EMF’s in our environment. From an evolutionary standpoint the main EMF mitochondrial are built to work with are sunlight but today this is no longer true. The fact that sepsis now kills 18 million people per year in a 4G world these links are incredibly important to follow to gain insight on how we should handle sex steroid hormones as we age in a 5G world. I have a sense the answers won’t be easy because the equation requires nonlinear thinking.
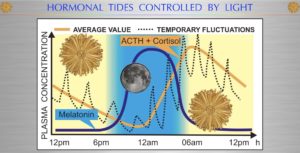
Your eye can be a clock or a camera. A blind man’s world is bounded by the limits of his touch and he relies on his timing; an ignorant man’s world by the limits of his wisdom; a successful man’s world by the limits of his vision and sense of timing. Man is the most complex eukaryote. Therefore he has the most sophisticated time piece in his eye that controls protein turnover. Protein turnover is a synonym for ubiquitin marking. Eukaryotes spend 80% of their total energy budget on protein synthesis.That process is controlled by ubiquitination rates in cells. This is why you need to understand ubiquitin. Once you master ubiquitin you can use that recovered energy to reverse illnesses. Each peptide bond requires 5 ATP to seal the bond. That amount is 5 times as much that is needed to polymerize nucleotides into DNA!! Each protein is reproduced in thousands of copies, which are continuously turned over by ubiquitin to repair wear and tear. Elevations of ubiquitin marking are usually associated with higher blood glucose and ammonia levels. This occurs because of damage in the urea and TCA cycles at Kreb’s bicycle. Cell become unable to use beta-oxidation and protein cycles for biosynthesis. Too many people are now blaming sugar and glutamine in cancer cases when it is clear the mitochondrial damage is making cancers have to use glycolysis and the PPP exclusively for bio-synthesis because of mitochondrial damage. Cancer is a circadian disease in my opinion.
Medicine today treats the eye as a camera almost exclusively, when its most important physiologic role is as an optical clock. It turns out cataract formation and glaucoma are the best evidence that the timepiece in your eye is no longer working in concert with your gut, skin, or any of your cells properly. I believe all tissues contain time crystals that go awry because of a lack of sunlight during the day and a lack of darkness at night. When this occurs diseases usually follow.
Breast cancer patient usually has very abnormally low Vitamin D and melatonin levels when they are sampled. They rarely are because conventional oncologist DO NOT believe cancer is a circadian disease caused by mitochondrial dysfunction in the mitochondrial matrix. They also do not understand how glucose fits into this story. Most people today view elevated blood glucose as a pathologic condition related to a Warburg metabolism. That is another big error. When the clock in your eye is altered there is downstream collateral disarray in the peripheral clock genes. There is now another way to perceive an elevated blood glucose as a clinical sign of elevated ubiquitin ratios in all proteins because of a dysfunctional TCA and urea cycle.
Glucose is fully capable of braking ubiquitin cycling when ubiquitin is coupled to the cell cycle by normal solar light cycles as the picture above shows, but not when it is uncoupled and isolated due to altered solar cycles. The reason glucose has this ability because it contains a strong blue light signal in it for the SCN in the eye to provide negative feedback for the SCN. This ability is lost when light cycles are uncoupled from the nitrogen cycle in the eye or gut. When it is isolated, glucose levels go through the roof BY DESIGN to stop the PER 1 and PER 2 clock genes from turning over proteins by ubiquitin marking in cells. I believe eventually the narrative that cancer feeds on glucose will die a fiery death under the weight of new data about the PER gene products. PROTEIN Turnover is the most energy costly activity a living thing does.
Most view the eye as a camera, but some of us see it as a optical lattice clock first. HYPERLINK
In my recent webinar series for members on my site, (March through June 2015) I taught you about how the loss of negative feedback control in coupled biologic systems is the sentinel event for aging and disease generation. Moreover, I showed you what happens when you lose it on one side of the coupled event. There I used predator or prey to make the point. If you alter the balance of predator or prey the result is always the EXTINCTION of both animals. I have told you that in aging and neolithic disease generation that NAD+becomes altered in relation to NADH. This occurs because of defects in the TCA and urea cycles where the hydrogen is recycled from metabolic substrates to the electron carriers used in glucose metabolism at cytochrome 1.
The chronic loss of NAD+is the critical sign of a loss of negative feedback control of the ubiquitin cycle. This data scales directly to our molecular circadian clock and our peripheral clock genes (CCG’s). The LCHF cancer researchers are way off on their beliefs about the demonization of glucose in most cancers. This same relationship also exists between the two coupled systems that control the eye clock protein timing mechanisms. When these proteins are drowned in blue light, it changes the molecular resonance coupling and this causes the extinction of the gears that control your timing mechanism in every system in your body. This is why blue light is associated with just about every neolithic disease known to man. Where it occurs first is where diseases manifest soonest.
The current model of the mammalian circadian clock includes two interlocking transcription-translation feedback loops comprised of several so-called “clock” genes and their protein products, which ultimately regulate the transcription of “clock-controlled” genes. These feedback loops consist of positive and negative components. The positive components include the basic helix-loop-helix-PAS domain transcription factors, CLOCK, and BMAL1. These transcription factors heterodimerize, translocate from the cytosol to the nucleus, and bind to circadian E-box promoter elements that enhance the transcription of genes encoding the negative components PERIOD 1 & 2and CRYPTOCHROME 1 & 2.
This month in May 2018: It appears my educated guess about PER1 and PER 2 in breast cancer was spot on. Read this hyperlink.
The CRYPTOCHROME and PERIOD proteins feedback inhibit the transcription of the Cryptochrome and Period genes by blocking CLOCK/BMAL1-mediated trans-activation. The second feedback loop involves the trans-activation of the Rev-Erbα and Rora genes by CLOCK/BMAL1. The protein products of these genes compete for binding to RRE elements in the Bmal1 promoter, driving a daily rhythm of Bmal1 transcription and closing the second feedback loop. Rhythmic expression of these clock gene products produces circadian clock outputs by regulating transcription of clock-controlled genes (CCGs). At least some of these CCGs, including aanat, the gene encoding the penultimate enzyme in the melatonin biosynthetic pathway, contain circadian E boxes, which have a core nucleotide sequence of CACGTG and are activated rhythmically by CLOCK/BMAL1. Post-translational regulation, including phosphorylation, acetylation, ubiquitination, sumoylation and proteasomal degradation is also important in the regulatory mechanisms generating the circadian oscillation. All of these coupled processes become unhinged from light signaling to affect nitrogen, water, and carbon flows in cells to cause many neolithic diseases because of mitochondrial dysfunction.
SUMMARY:
Once your SCN timing mechanism goes haywire it is a matter of time before your circadian clock genes in tissues the SCN controls also goes haywire and result in diseases. What will be the ultimate result? EXTINCTION of both sides of the circadian timing mechanism and cancer is the ultimate result. Mitochondrial are self-regulated by just programs in humans. Those programs are autophagy and apoptosis. Melatonin helps augment autophagy by repairing mtDNA and acting as an antioxidant in darkness. Interestingly enough, it is made first in the eye by an aromatic amino acid by sunlight. Apoptosis is controlled by UV-A light because it releases nitric oxide which inhibits ECT at cytochrome 4 to increase cell suicide. Cancer cannot exist with intact apoptosis. This makes me believe sunlight is the ultimate vaccine for cancer. The key is your skin and eye have to sense the full spectrum of the sun’s light to get the benefit. It should be interesting to you that plants do not get cancer in nature. There is a deep reason for this that will be covered in this series. Cataracts, glaucoma, and many autoimmune conditions are earlier appearing neolithic diseases that often predate oncogenesis because of an elevated ubiquitin rate which is a MARKER for matrix dysfunction that cause abnormalities in NAD+.
CITES:
1. Herzog ED. Neurons and networks in daily rhythms. Nat Rev Neurosci. 2007;8:790–802.
2. Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, et al. Resetting central and peripheral circadian oscillators in transgenic rats. Science. 2000;288:682–685.
3. Ko CH, Takahashi JC. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;2:R271–277.
4. Munoz E, Baler R. The circadian E-box: when perfect is not good enough. Chronobiol Int. 2003;20:371–88.
5. Gatfield D, Schibler U. Proteasomes keep the circadian clock ticking. Science. 2007;316:1135–1136.
The title is provocative because contrary to popular belief a high fat diet can make you quite obese when a couple of variable are present that our modern environment favors today.
This blog fully explains why I knew I had to quit being on call and why I had to regain my time at night and weekends to remain well as I age and am afflicted by mitochondrial damage built into us by nature. If I did not test my own biases I would have be facing different challenges then I do today.
THE BLOGS MAIN POINT UP FRONT:
Focusing in on food is losing proposition for the aging human. This sound nuts when you first hear it but is very wise once it is fully explained so you can understand the perspective.
Black Swan mitochondriacs have learned this lesson the hard way. They focus in on the engine and not the fuel to the engine. The analogy is simple. If you want your Ferrari to go 225mph constantly the main thermodynamic variable that will help you attain this goal is making sure the engine works perfectly. If the engine is perfect it can consume 87, 90, or 93 octanes to get this goal at any one time. If you use cheap gas over time it can affect the performance of the engine, IF YOU NEGLECT to maintain the engine maintenance. This brings up an interesting question to think about before I go full nuclear on the science.
Why do so many gurus recommend a high-fat diet to people who are aging, people with mitochondrial damage, or people with mitochondrial diseases?
Is ketosis supposed to be from the foods we eat of the liberation of fat from our own fat mass?
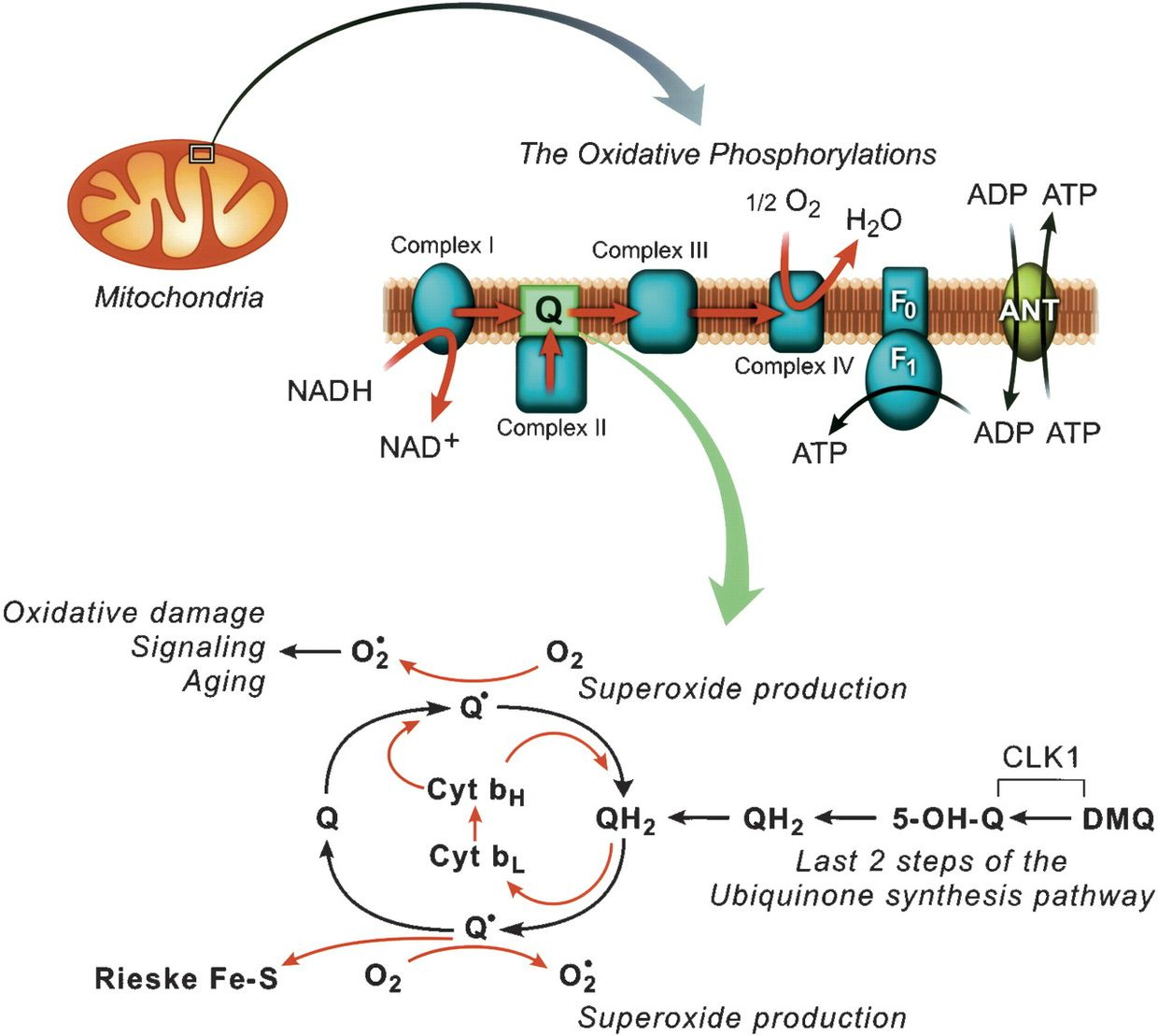
What good is a ketogenic diet from fat if you cannot use it because your matrix is defective? The only way to use fat is via beta-oxidation via the TCA cycle. Oxidative phosphorylation is made possible by the close association of the electron carriers with protein molecules along the inner mitochondrial membrane. The proteins on this membrane are unique, devoid of DHA (a special PUFA), and seem to guide the electrons along the respiratory chain so that the electrons move sequentially from one enzyme complex to another—with no short circuits in normal functioning.
This leads to a stable ROS signal in mitochondria that build complexity using information quanta in electron and proton quantum spin. The ROS signal is a function of oxygen content and the electrons within the inner mitochondrial membrane NOT experiencing any electrical short circuits. Does this situation persist in life? No it does not, it varies as the distance between the proteins change and this is what aging really is. We call this heteroplasmy in biology. Short circuits become more likely and this changes the free radical signals possible while lowering oxygen levels which indirectly forces NAD+ at cytochrome 1 lower.
NAD+ stands for nicotinamide adenine dinucleotide.It has two states, one with electrons and one without.The electrons come from carbohydrates that grow in powerful light latitudes.
Moore circa 2010 sans “massive” technology diet
Moore (2017) on nutritional ketosis running his empire on social media
It has been widely assumed that a ketogenic diet using ketone bodies as a substrate can raise endogenous NAD+ to improve redox status but all these studies were done on nocturnal mammals who have scotopic retinas and skin. This is a very bad assumption considering that NAD+ is a FLUOROPHORE protein with a 340 nm spectral pattern. UVA light makes nitric oxide in our skin, blood vessels in our tissues to affect mitochondrial function.
This is why nature built it to handle excited electrons from foods that grow when UV light is present to a high degree. This error has spilled over to the LCHF and ketogenic groups in humans. For nutritional ketosis to work it has to be coupled with UVA exposure of the eye and skin because it lowers ECT and energy production in the mitochondrial matrix.
If it is done under the power of blue light, the ROS from melanopsin signaling destroys the quantization of NAD+ and NADH at cytochrome 1 to make is highly dysfunctional (picture above). This means mitochondrial damage changes the kinetics of how NAD+ can operate in HUMANS irresepctive of our dietary fuel choices. This means dietary fats can make us quite fat if cytochrome is damaged and IR-A and UVA light are absent from our life.
The recent research cited below in cite 1 (pic above) shows these assumptions might be dead wrong in humans because it appears that the ratio of NAD+ and NADH can vary independently because they are compartmentalized.It turns out NAD+ and NADH are affected by the PHYSICAL location of certain metabolic pathways.Different locations introduce timing and relativity to the equation, and no one seems to realize how this is affected by altered by the external lighting the animal is under to change the kinetic flux of the pathways mentioned in this blog.
For example, the TCA cycle is in the matrix, while the urea cycle is in the cytosol. The place where they meet is called Kreb’s bicycle in research circles. Higher quality research is needed to further identify where and when ketogenic therapy increases the NAD+/NADH ratio in humans. The study needs to be done under both fake light and sunlight to see the real effect on human obesity because of the absorption spectrum in the inflection point of the UVA and UVB part of the visible spectrum of light.
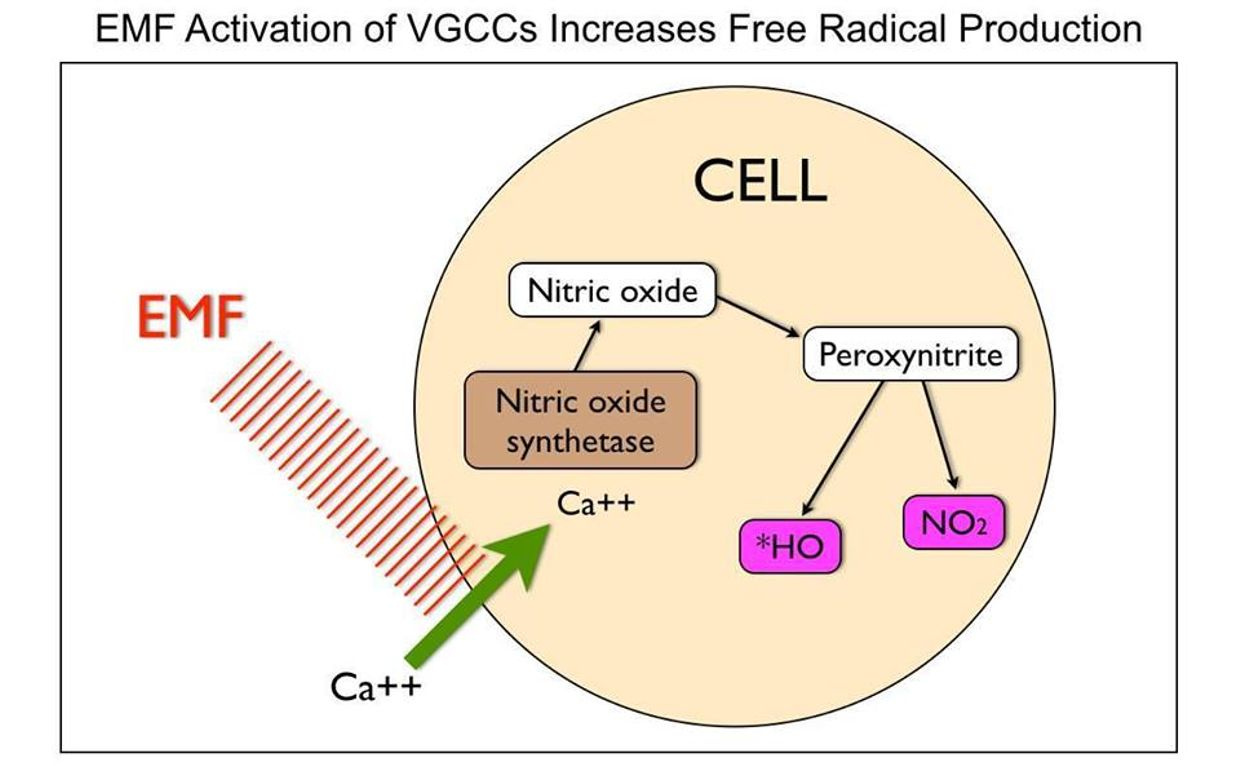
THE INCIDENT EMF CHANGE THE FREE RADICALS MADE IN THE MITOCHONDRIA.
UV-A light makes Nitric oxide (NO). Nitric oxide inhibits electrons motions from cytochrome 3 to 4. It is a breaking mechanism to keep us thin and if you do not get any UVA and replace it with blue light from technology you just gave your body the signal to FATTEN.
These ideas will be critical in delineating specific downstream effects. This implies that a ketogenic diet could be highly fattening to humans who are blue light toxic. Blue light toxicity leads to ROS in the matrix.
There are two type of blue light Hazards known in humans. Noell et al. has described a rhodopsin mediated class 1 blue light hazard (BLH) and Ham et al. categorized a class 2 blue light hazard mediated by lipofuscin. Each cause different collateral effects because of the metabolic pathways they destroy.
Right now I believe the observation of modern humans supports this position fully and this 2018 paper from UT tells me the things I wrote in the Ubiquitination 4 and 5 blogs has even more wind in those sails than I realized when I wrote them years ago. The last cite below shows you how long I have believed that something else beside insulin was behind fattening of humans in technology 24/7.
It will be critically important in future studies to optimize the methodology carefully to ascertain if changes in the NAD+/NADH ratio are caused by changes in NAD+ or NADH solely (unlikely), or as levels of these two redox molecules can also vary independently. This is more likely because of the circadian mechanism at play at cytochrome 1. Supplement makers are now heavily pushing NAD+ supplements because they are betting that raising NAD+ alone varies by its concentration. That is unlikely because its effect is now known to be compartmentalized and is subject to pathway kinetic effects and flux.
Many low carb researchers/gurus have proposed that the decreased reduction rate of NAD+ to NADH during ketone-based metabolism increases the availability of NAD+ and thus alters the NAD+/NADH ratio.There are several problems with this assumption. The first one is that NAD+ is reduced by the addition of hydrogen to make NADH. This happens in the matrix which is a tightly regulated organelle. The hydrogen in this location must come from the hydrogen pool in the matrix. This is not true in all tissues in humans as the picture below shows.
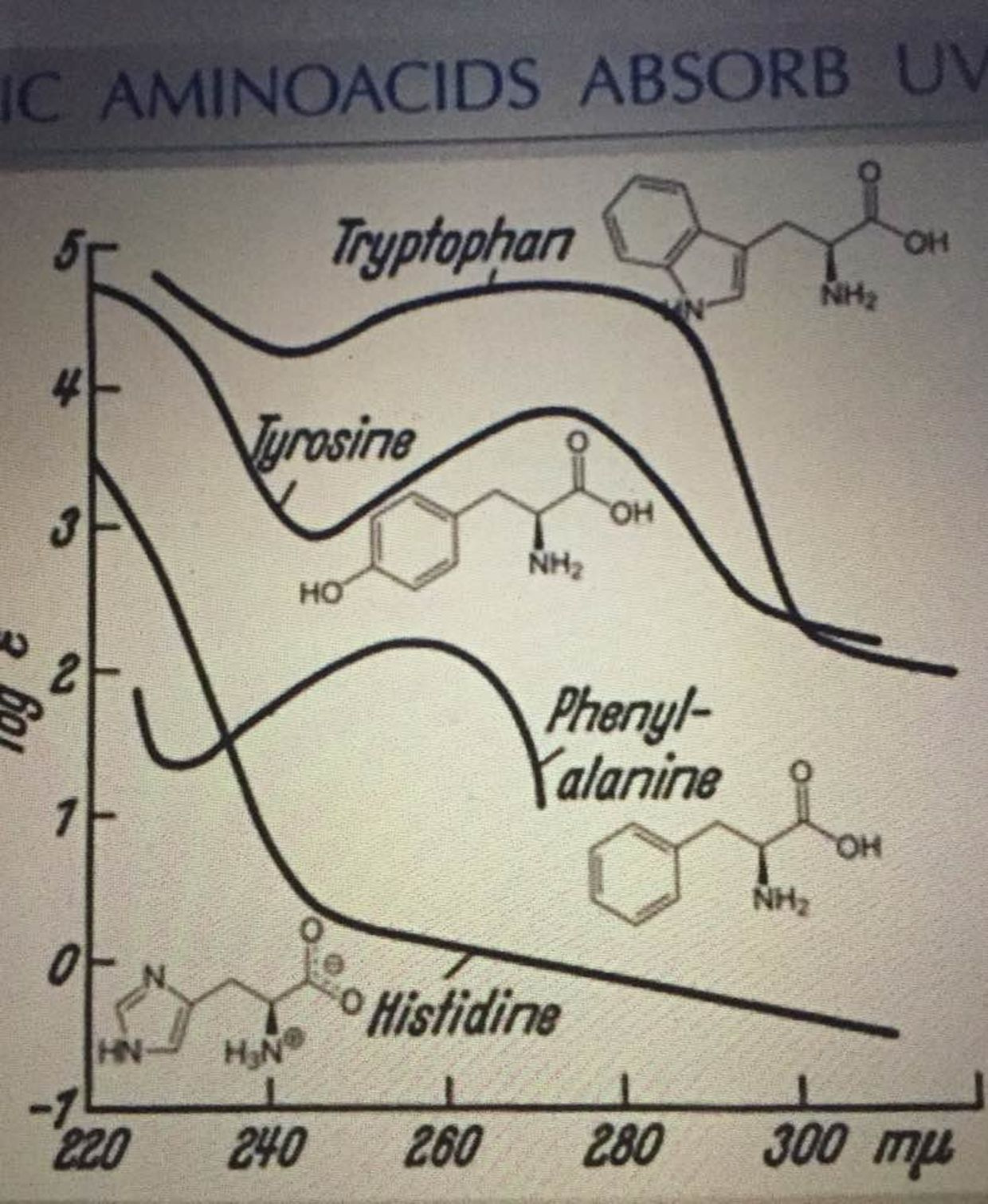
For example, the heart and kidney can use alternative pathways to create NAD+. For example, the de novo synthesis (the deamidated pathway) uses the amino acid tryptophan, which is metabolized to form biosynthetic intermediates. These intermediates ultimately generate nicotinamide (the pyridine moiety of NAD+) and then form NAD+. Another pathway to NAD+ is the use of dietary vitamin B3 compounds, including nicotinic acid, nicotinamide and nicotinamide riboside, serve as NAD+ biosynthetic precursors and are salvaged from the diet (the amidated pathway) to generate cellular NAD+.
Tryptophan works with UV light from 260nm-290nm as the picture below shows. This is big time short wave UV light close to the UVC range. Why doesn’t anyone see these connections?
NAD+ is a hydride acceptor from the TCA cycle that forms the reduced dinucleotide NADH. The NAD+/NADH nucleotide pair is vital for driving reduction-oxidation (redox) reactions in energy production to oxygen in human mitochondria. Furthermore, NAD+ is a precursor for the phosphorylated dinucleotide pair NADP+/NADPH, which is required for several cellular biosynthetic pathways (Pentose phosphate pathway =PPP) and to protect cells from reactive oxygen species (ROS) made in the mitochondrial matrix. So when NAD+ is lowered for any reason, the quantum clinician knows that the matrix is making more ROS/RNS than it should be while liberating more ELF-UV as a result.
This raises another question, where does the ELF-UV light come from?
That answer will be covered soon the QT series here and in my Vermont 2018 talk on June 1.
NAD+ is the oxidized version of cytochrome 1 meaning it is lacking an electron from dietary carbohydrate breakdown.NADH is its reduced form of this chemical, meaning it has these electrons from carbohydrates.Together both make up the thermodynamic couple that carries electrons from carbohydrates in humans.NAD+ is part of the cytochrome 1 couple that carries carbohydrate electrons to oxygen in normal mitochondrial function.It is afluorophore-proteinthat has an absorption spectrum of 340nm which is also deep in the UV range.
KEY BLOG POINT:
The team of researchers at the University of Texas from cite 1 below has found NAD+ synthesis and consumption integrate glucose metabolism and adipogenic transcription during adipocyte differentiation. In their paper published in the journal Science, the group described their research into how glucose is converted into fat in the body. As obesity rates continue to climb around the globe, these scientists wanted to explore why obesity is rising as NAD+/NADH couple in mitochondria is defective. None of them realize that nnEMF cause AMPk amplification and more glucose to be spilled in the blood because cytochrome 1 is destroyed by the frequencies of light used by modern communications. The defect can be enhanced by blue light toxicity in the skin and eye because as melanopsin is ruined, melatonin is lowered and we cannot maintain our mtDNA to keep the cytochromes in ECT working well or replace them. UV sunlight helps us eat less by the design in our mitochondria.
They do not seem to realize that UVA light from the sun has to excite these electrons to get the effect. Just eating tremendous amounts of them does nothing but cause them to go to storage via the NAD+ PPP pathways. They also do not seem to be aware that the H+ need to make NADH whole again must come from the mitochondrial matrix.It cannot be in any other isoform of hydrogen. The May 2018 webinar laid the foundations out for you.
If these light conditions are NOT met on the skin and eye, NAD+ will remain abnormal.This is why blood glucose rises. The electrons cannot be fed in if cytochrome 1 is nonfunctional. As blood glucose rises, insulin secretion increases as a collateral issue from beta cells and this causes the fat mass to expand. The cause of diabetes is really mitochondrial dysfunction and not an insulin driven mechanism. This is why the UT researchers remain confused. They do not realize where the pieces fit.
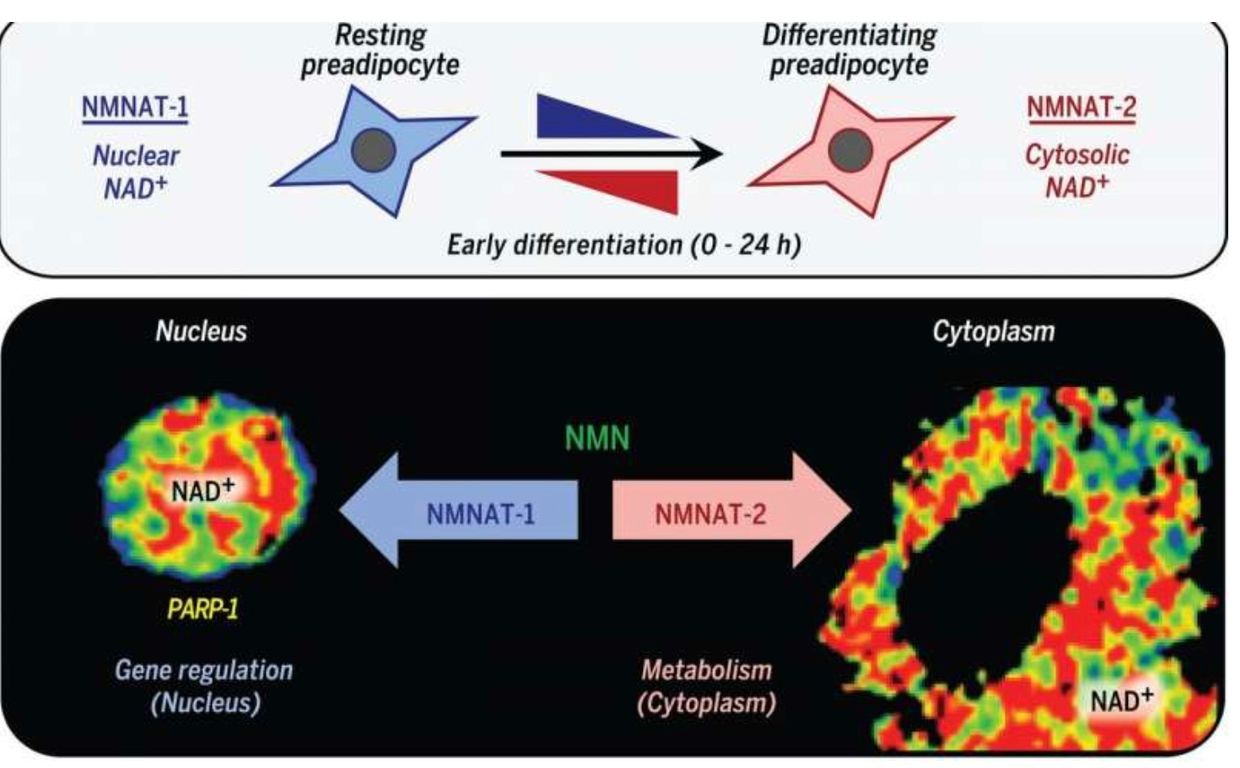
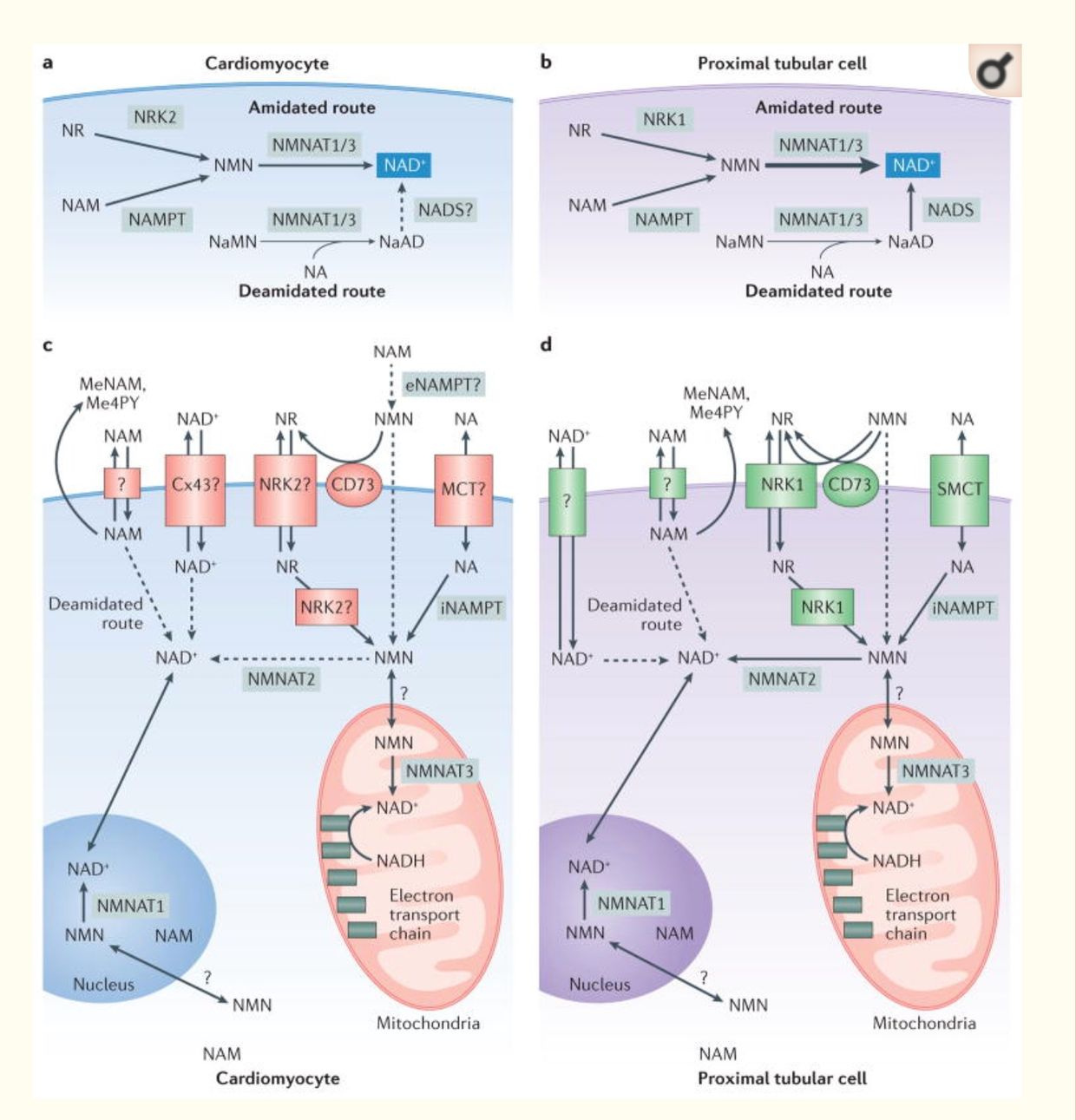
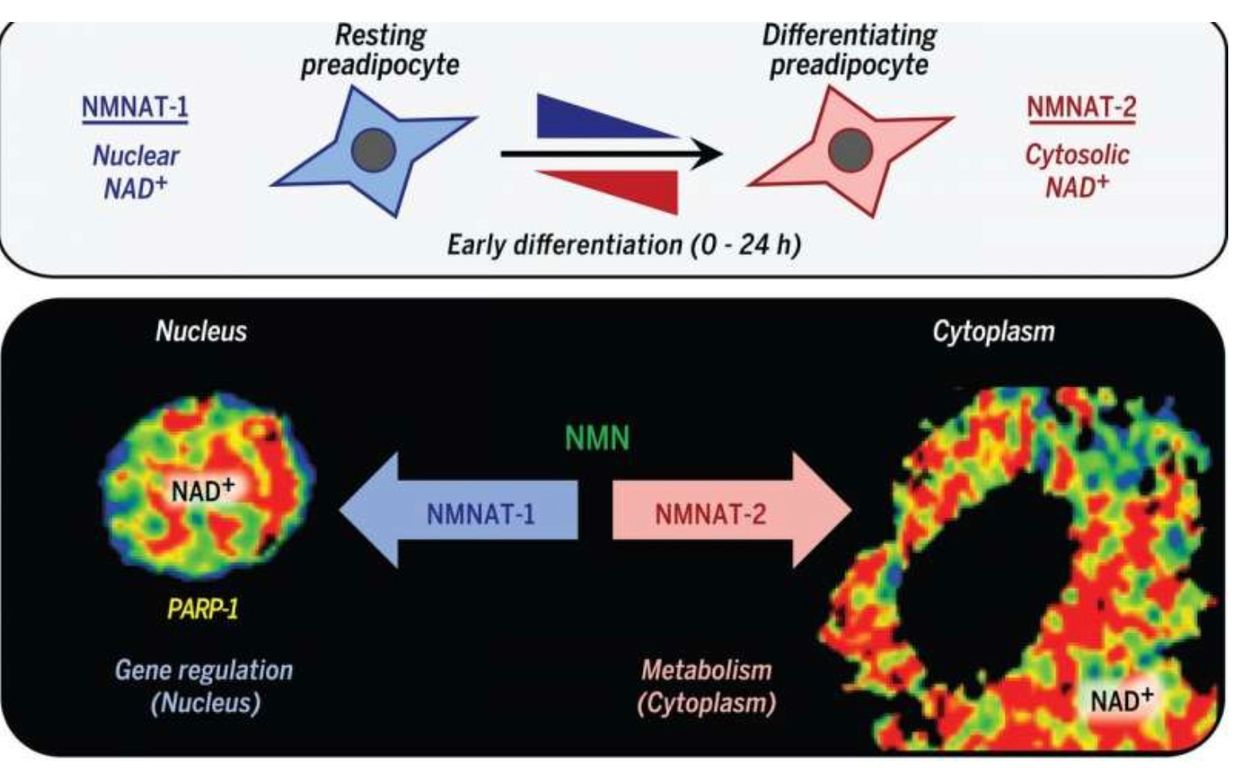
NAD+ synthesis is compartmentalized and within several membrane-bound organelles associated with the outer mitochondrial membrane via the Tensegrity system.It is synthesized in distinct subcellular compartments by three different synthases called NMNAT-1, -2, and -3 as the picture shows.
KETOSIS IS SUPPOSED TO BE USED FOR OUR OWN FAT OR IN WINTER, NOT 24/7.
We are designed to use our OWN fat in the subcutaneous space in autumn and winter to run these programs when UVA and UVB light are absent. Eating fat is not the same idea.
Eating a high-fat diet is one way to raise NAD+, but eating this way poses a risk if the mitochondrial colony in that person’s tissues cannot effectively use beta-oxidation of the TCA cycle.In this case,the nutritional ketosis drives an increased adipogenesis via adipogenic signaling that is rapidly induced by the cytoplasmic NMNAT-2.
This isoform of the synthetase competes with a nuclear version calledNMNAT-1 for the common substrate, nicotinamide mononucleotide.This chain of events leads to a precipitous reduction in nuclear NAD+ levels.This signal inhibits the catalytic activity of poly-adenosine diphosphate (ADP)–ribose] polymerase–1 (PARP-1).This is an NAD+-dependent enzyme that represses adipogenic transcription by ADP-ribosylating the adipogenic transcription factor C/EBPβ. So when somebody advocating a high-fat diet in the face of mitochondrial damage due to a chronic non-operational cytochrome one, you can get quite fat from eating fat.
This is especially true when the TCA cycle is dysfunctional for any reason. Blue light toxicity cause breakdown of the outer mitochondrial membrane and this allows deuterium to enter the TCA and urea cycle. This destroys the kinetics of the NAD+/NADH couple. It has many more collateral effects. Most people in the LCHF community have no clue this mechanism exists because they do not read mitochondrial papers carefully.In fact, the reversal of PARP-1–mediated repression by NMNAT-2–mediated nuclear NAD+ depletion occurs in response to adipogenic signals which drives adipogenesis in humans.
This is the dominant way technology use causes obesity via the melanopsin signaling in the eye, skin, and fat.I call this the Jimmy Moore effect.Why?
As NAD+ drops because of its own metabolic consumption, there is a serious loss of information transfer in mitochondria. This is why low NAD+ levels are associated with aging and obesity and many other chronic diseases. If your TCA and urea cycle are broken, oxygen levels also drop. This drop in oxygen means the cell CAN ONLY use glycolysis and the PPP to drive biosynthesis.
IS NAD+ AFFECTED BY THE PPP?
Yes it is.
NAD+ is broken down into nicotinamides and ADP-ribose that feed into the PPP. Glycolysis and the PPP are older metabolic pathways involved in cell biosynthesis that operate in lowered oxygen environments with poorer redox capability. These two pathways also get pulled into double duty when the TCA or urea cycle are non-functional because of the blockade at fumerase. Fumerase is where both of these cycles meet at the cell membrane. This situation is usually caused by cell membrane damage in the mitochondria which unleashes deuterium in the matrix where these two pathways meet one another. These two pathways were favored by the first two kingdoms of life prior to the Cambrian explosion and early after the Cambrian explosion before atmospheric oxygen spiked for mitochondria to take full advantage of the electron negativity to pull electrons with some force toward oxygen. This means that NAD+ must be resynthesized in some fashion FAST enough for a normal cellular function to continue. Many researchers have noted that some prior papers have suggested that lower-than-normal levels of NAD+ can alter metabolism, leading to higher disease susceptibility.
Is there another way to help NAD+ levels when the TCA and urea cycle are functioning poorly?Yes.We can use polyphenols that affect the sirtuin axis. This is why I am fond of high polyphenol wines from high altitudes. They have two beneficial effects on hydrogen for the NAD+/NADH couple. The second way they operate is that these grapes are grown with deuterium depleted rainwater at higher altitudes and latitudes. I have found some coffees can do this as well and will be talking about them in the future. In particular, NAD+ functions as a co-substrate in deacylation reactions which are carried out by the sirtuin family of enzymes. These NAD+-dependent deacylases control several aspects of metabolism and a wealth of data suggests that boosting sirtuin intake may affect the endogenous activity NAD+. The mitochondrial sirtuins control metabolism and information transfer in the matrix by coupling NAD+ consumption with deacylation on critical lysine residues of metabolic proteins in the mitochondria, thereby regulating flux through cellular metabolism This requires the use of some salt and a higher saturated fat diet regimen loaded with seafood and pork. Soon you’ll find out why this is the case here on Patreon.
CITES:
1. Keun Woo Ryu et al. Metabolic regulation of transcription through compartmentalized NAD+biosynthesis, Science (2018). DOI: 10.1126/science.aan5780
2. Achanta L. B., Rae C. D. (2017).β-Hydroxybutyrate in the brain: one molecule, multiple mechanisms. Neurochem. Res. 42, 35–49. 10.1007/s11064-016-2099-2
3. Ahn Y., Narous M., Tobias R., Rho J. M., Mychasiuk R. (2014). The ketogenic diet modifies social and metabolic alterations identified in the prenatal valproic acid model of autism spectrum disorder. Dev. Neurosci. 36, 371–380. 10.1159/000362645
4. Belenky P., Bogan K. L., Brenner C. (2007). NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19. 10.1016/j.tibs.2006.11.006
5. Boison D. (2017). New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 30, 187–192. 10.1097/wco.0000000000000432
6. Bough K. J., Rho J. M. (2007). Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48, 43–58. 10.1111/j.1528-1167.2007.00915.x
7. Bough K. J., Wetherington J., Hassel B., Pare J. F., Gawryluk J. W., Greene J. G., et al. . (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. 10.1002/ana.20899
8. Braak H., Braak E. (1998). Evolution of neuronal changes in the course of Alzheimer’s disease. J. Neural Transm. Suppl. 53, 127–140. 10.1007/978-3-7091-6467-9_11
9. Branco A. F., Ferreira A., Simões R. F., Magalhães-Novais S., Zehowski C., Cope E., et al. . (2016). Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur. J. Clin. Invest. 46, 285–298. 10.1111/eci.12591
10. Brownlow M. L., Benner L., D’Agostino D., Gordon M. N., Morgan D. (2013). Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS One 8:e75713. 10.1371/journal.pone.0075713
11. Brownlow M. L., Jung S. H., Moore R. J., Bechmann N., Jankord R. (2017). Nutritional ketosis affects metabolism and behavior in Sprague-Dawley rats in both control and chronic stress environments. Front. Mol. Neurosci. 10:129. 10.3389/fnmol.2017.00129
Can A Cocktail Of Vitamins And Steroids Cure A Major Killer In Hospitals?
Sure it can because that cocktail mimics the ketogenic diet. In a 5 G world this might be the most common serious cause of death I expect to see spike in the next 5-7 years.
Both ideas cause a recycling of cell water in the cytoplasm. I don’t believe keto is a weight loss Rx at its core either. It is something rather different that mimics the effect of Vitamin C in cleaning the TCA and urea cycle of deuterium at Kreb’s bicycle. Because of the shape of our teeth and the shortening of our gut humans lost their requirements for Vitamin C but it raised our needs for marine fats, iodine, and animal fat. This fostered a seasonal ketosis that turned over the cell water in the matrix.
Ketosis is a novel seasonal way to drive matrix water replacement while autophagy can control cell membrane turnover and the movements of deuterium. In sepsis, both autophagy and apoptosis are uncoupled because of defective enzyme kinetics at fumerase, so this acutely lead to multiple organ failures quickly and causes a quick death.
It would be wise for hospitals to use red light in these patients rooms at the same time but they do not appear to understand that the 4th and 5th cytochrome react briskly to red light even when ECT is broken. This could be extended if windows were opened in an ICU to allow UV-A light in to patient room to help re-establish apoptosis. Another helpful adjunct would be the use of IV DDW. To date no one has tried this in the literature as far as I can tell.
This is so powerful, that in some cancers ketosis can exhibit synergistic antitumor activity and preferentially kill tumor cells by autoschizis, a novel type of necrosis characterized by exaggerated membrane damage and progressive loss of organelle-free cytoplasm through a series of self-excisions. During this process, the nucleus becomes smaller, cell size decreases one-half to one-third of its original size, and most organelles surround an intact nucleus in a narrow rim of cytoplasm. This narrow rim is the key marker for extreme turnover of cell water.
The key in sepsis and cancer is to change the fractionation of hydrogen in the cytosol. In sepsis, the cytoplasm becomes acutely overloaded with deuterium, but in cancer, it is a chronic change. In both cases, the cytoplasm is loaded with the wrong type of hydrogen isotope used in the urea and TCA cycle. While the mitochondria are condensed, tumor cell death does not result from ATP depletion.
Many people have heard me use the term zip code in discussing quantum biology, but no one has ask me what I mean by it. What is “zip code” to you? I believe our zip code is the quantum fingerprint in nature we resonate with PROPERLY. Each hour of every day at every location has its own distinctive color, a family of frequencies, and a particular odor. Some of the smallest details inside of cells pay attention to these things. Nature always provides a solution, and our mitochondria was built how to solve the cipher. Our senses have to be blinded to the mystery. We can overcome that haze of understanding only if we know where to look for the answers. Life provides a myriad of vibrations but some resonant with us more than others. That is our connection to the whole. If we only live through the eyes of the others and don’t try to capture the vibration of our own individual experience, we may miss out the beam and brilliance of authenticity that is essential to move forward in nature. Allow no one and no thing to cause a schism between you and your intuition. Disorder creates connections──and that is the resonance life is built on.
ENTROPY = disorder. Resonance creates an order from the disorder. The probabilty of things in us and things in the sun and Earth cause a connective coupling called a correlative novelty. ‘Correlated novelties’: are a big thesis in how entropy drives the arrow of time in quantum thermodynamics. The more entropy we dissipate the more time become irreversible. This is why we perceive life as we do.
HOW DOES THIS HAPPEN?
In life, as everywhere in the cosmos there exists a struggle between in matter, both biotic and abiotic. Chaos is entropy in abiotic systems and it turns out entropy appears to organize biotic systems without energy either. (represented by fear in biotic systems) Entropy appears to lead to an order which can organize into a zero entropy state of matter than keeps life far from equilibrium. If there’s a kind of physics behind biological teleology and agency, it must have something to do with the same concept that seems to have become installed at the heart of fundamental physics itself: information. Biophysics is as much about how mitochondrial create the energy flux as it is about how it processes environmental information accurately. To extract meaning from information or from energy something has to know how to do it. How does this happen? It appears this information in cells is harnessed using water’s shape shifting liquid crystalline abilities under solar powers direction. According to thermodynamics, the capacity to extract useful work from the energy resources of the universe is always diminishing.
A mitochondria is ‘demon’ who deals in information and heat processing and energy flux. First, for the demon to dominant, it always must have more information than we do about the environment. The organism housing this colony of mitochondria must be blinded to this information and energy. This is just another facet of the quantum Zeno effect we see in the retina and skin with respect to UV and IR light. Humans cannot see either of them, because if they could they could not use either frequency family to regenerate their tissues via autophagy.
Our colony of mitochondrial demons must be able to see or sense all of the molecules individually, rather than just statistical averages of H+, deuterium content, and excitied electrons on the inner mitochondrial membrane. And second, the colony of demons within us has intention: a plan to separate the hot from the cold using uncoupling proteins to make water liquid crystalline. By exploiting its knowledge with intent, mitochondrial demons can defy the classical laws of thermodynamics by usng quantum thermodynamics
There is a deep connection between thermodynamics and the processing of information — or in other words, is related to the computations that mitochondria must make using the quantum spin state of electrons and protons. Mitochondria make free radicals that have unpaired electrons with a unique quantum spin state. The matrix excludes deuterium while the blood plasma is filled with it. There seems to be an organizing process present because of the spin state of subatomic particles that is a hidden variable that limits biologic understanding.
Mitochondria are electron and proton ciphers and accountants for these things. This happens to be why the leptin receptors in the hypothalamus are loaded with mitochondria. Rolf Landauer showed that even if the computational aspects of an information demon can gather information and move a frictionless door with no seeming energy cost in his experiments. He proved Maxwell’s ideas about the second law was correct.
Landauer also pointed out a penalty must eventually be paid back to nature. He showed that no mitochondrial demon can have unlimited memory of every molecular motion, unless it wipes its memory clean every so often. This raises the question, is this why sleep and mitochondrial function are ubiquitous in living things?
The best two ways to improve sleep are to observe the sunrise and block our eyes and skin from man made light once the sun sets.
Is this why poor sleep and mitochondrial diseases are linked? I believe it is. All quantum computational demons must occasionally wipe its memory clean — forget what it has seen and start again — before it can continue harvesting energy and information simulataneously. This night day on off switch is likely why most of the biologic cycles are feed by the sun’s energy and information of night and day. This act of information erasure has an unavoidable price: It dissipates energy, and therefore increases entropy. And that increase of energy does not go to waste in living systems. We use the increasing entropy to ORGANIZE information we collect.
It seems to me living quantum systems are expert in the information sharing side of the equation to organize and minimize chaos using biomolecules from the mitochondria to other parts of the cell, while extracting massive amounts of information about the universe they exist in. Two groups of researchers found in 2017 that the quantum information describing the particles’ energy and quantum spin states can act as a kind of currency that enables trading between the reservoir’s energy and angular momentum supplies.
This has a big implication for mitochondriacs because this organelle deals in deciphering and creating the quantum spin of electrons and protons and the information and energy they contain. The deciphering and creation of spin varies as the power densities of seasons changes as the planet moves around the sun. Electrons have quantum spin and they have information about the seasons in the power of photons that excite them. The spins of electrons can be altered in mitochondria to make free radical signals which can be used to drive decision making processes inside of cells. This will alter biochemistry within the cell. The quantum spins of protons also can be sensed by the mitochondria because the mitochondria generates a magnetic field and H+and D have two separate magnetic moments that will act differently within the same magnetic field. This difference is like a binary change in a computer that can be used quantum computation to decipher the information contained within the message.
More over, protons spin is also sensed by the mitochondria because all the channels in the mitochondrial matrix are quantum nanomachines that are built exclusively for H+ and not for the larger atom of deuterium. The level of deuterium in the matrix therefore also simulates the season inside the matrix of the mitochondria. This alters the metabolic rate of the mitochondria as well. All of this is programmed by light and is how ZIP CODE is coded for inside of us.
The more deuterium that is present, the more swelling one should expect. This shows you why thermodynamics and size and shape changes within the colony of mitochondria in tissues can be sensed macroscopically by our sensory systems in our thalamus. This alters the Tensegrity of the system. The human thalamus also has normal resonant frequencies it is tuned too. That frquency is the Schumann frequency of the Earth. Think of it as the electric and magnetic heart of Earth that is communicating with your thalamus.
Living quantum systems achieve a non equillibrium state, by capturing and storing information in our subatomic particles through the entrie system. Mitochondria are the key part of cells that are capable of doing these computations using free radical creation and the movements of hydrogen to build organization in a cell. The nucleus cannot do these computations in eukaryotes. Mitochondria offload most of the information to the nucleus by changing small poteins made by mitochondria carrying data to change the activity of the nuclear genome. This information is encoded and stored magnetically in their nuclear genes and passed on from one generation to the next via the germline.
This is a set of instructions for reaping a non equilibrium state by using the negative entropy built around the quantum spin states of electrons and protons. This is why most repicative structures in nature offload deuterium to their seeds and offspring. It can be used to anchor points in the organism like a thumb tack to align the body plan in the immature state in space and time using magnetic energy as the glue. Deuterium has a stronger magnetic moment and it also has a stong kinetic isotope effect to act as a buttress and trusses in body plan construction in cell membranes. The added mass allows the blueprint to drive all growth and metabolism into the adult form as excited electrons are pumped into the system and trapped by mitochondrial mechanism to build complexity. Mitochondria and DNA are examples of aperiodic crystals that go by another name called liquid crystalline quasicrystal. These are more time crystals in us that resonate with the sun and Earth vibrations. How they vibrate changes as the environment changes. Cells are designed to keep the nuclear genes tightly coiled and this changes what they can sense. This is akin to how the fret board works on a guitar. As fingers compress a string the frequency changes. This change alters signaling throught the time crystals. All of this is transferred over protons in water to all parts of the organism. The exclusion zone of water is life’s main crystal. This crystal has a different physical characteristics when light hits water made in a mitochondria (DDW).
AM sunlight has a specific resonance for EZ water. It is blue and red light vibrating together that programs water. Artificial blue light from tech screens changes the resonance in water. This results in a changing free radical signal in the central retinal pathways of the retina and can lead to mitochondrial damage in the pathways that lead to the central clock mechanism in the SCN. It is called the blue light hazard and well known. There are two types of blue hazard caused by different frequencies of blue light which changes proteins in the retina. Blue light also lowers ocular melatonin in the central retinal pathways and this lowers the efficiency of autophagy and apoptosis.
UVA increases melatonin and nitric oxide (NO) in blood vessels in skin and retina. NO inhibits ECT of mitochondria and this increases apoptosis to get rid of pre-cancerous cells with poor time crystals at Kreb’s bicycle. How? NO inhibits the flow of electrons in ECT and this is the signal that cytochrome 4/cardiolipin complex need to allow apoptosis. Cancer cells must inactivate apoptosis so they can become immortal as I laid out in the May 2018 webinar. Blue light thins the retina because of the altered free radical signals generated in the damaged mitochondria and this does not allow a normal NO signal to make other signaling molecules. To get NO and melatonin you need AM UV-A and IR-A from the sun. These frequencies improves autophagy and apoptosis via resonance.
Examine this graph. The sun hasn’t changed much in 4 billion years but screen use and blue light exposure certainly has. This is how a change in light zip code can ruin your ability to see.
It turns out discrete time crystals (DTC) have another characteristic that will interest you. In physics they are forbidden to occur in equilibrium situation. Life is built far from equilibrium.
The discovery of time crystals might sound pretty abstract, but it heralds in a whole new era in physics – this is going to help quantum biology supplant the old guard in biology. Darwinian evolution is now on a death watch. For decades we’ve been studying matter that’s defined as being ‘in equilibrium’, such as metals and insulators. But it’s been predicted that there are many more strange types of matter out there in the Universe that aren’t in equilibrium that humans haven’t even begun to look into, including time crystals. It turns out all living systems are made of these things. And now we know they’re real. The fact that we now have the first example of non-equilibrium matter will change perspectives on what is possible in biology. It will lead to breakthroughs in our understanding of the world around us, and the quantum process biology inside of us, as well as new technology such as quantum computing. Mitochondriacs will be on this leading edge.
The current definition of a crystal is based on the currently known catalogue of periodic and aperiodic crystals. Scientists currently do not know of any materials that have aperiodically ordered structures beyond incommensurate crystals and quasicrystals. The definition of a crystal also reflects the lack of understanding of what constitutes order in matter, and in this sense should be seen as a working definition that may well need to be revised in the future. In crystallography, order is linked to diffraction, which makes sense because diffraction is the method of choice to experimentally determine the structure of a solid.
The human brain is very skilled at detecting patterns and recognizing order in a structure, and ordered structures permeate cultural achievements of human civilizations, be it in the arts, architecture or music. The ability to detect and describe patterns is also at the basis of all scientific inquiry.
In 2009, Sharon Glotzer and her group at Michigan discovered that entropy, a concept commonly conflated with disorder, can actually organize things!!! Her group’s simulations showed that entropy drives simple pyramidal shapes called tetrahedra to spontaneously assemble into a quasicrystal — a spatial pattern so complex that it never exactly repeats.
Water also uses these rearrangments when sunlight hits water in our tissues like blood via our skin.
It appears life’s time crystals is built from disorder to give all living things its arrow of time. Our eyes, skin, and gut are examples of time crystals. How ironic, but fitting, considering the counterintuitive nature of nature’s use of quantum processes.
It appears that cells have figured out how to utilize these queer processes close to 4 million years ago. It looks like water was the stage the show was built upon. The vibration of proteins stable on early Earth then began to vibrate with water and sunlight and the process of evolution began. It wa smillions of years before all the kinks were worked out in DNA and RNA mechanisms to select the proteins that resonated with the light that fell from the sun.
This discovery was the first indication of the powerful, paradoxical role that entropy plays in the emergence of complexity and order in all forms of matter. This principle is not just isolated in abiotic atoms. It appears in biotic systems too. Hemoglobin and porphyrins are well known biologic liquid crystals. Crystals are paradigms of ordered structures. Crystals are capable of doing amazing things to light rays from the sun or re-radiated light rays from other crystals in cells. While order was once seen as synonymous with lattice periodic arrangements, the discoveries of incommensurate crystals and quasicrystals has led to a more general perception of crystalline order, encompassing both periodic and aperiodic crystals. The current definition of crystals rest on their essentially point-like diffraction.
Cells use biophysical levers contained in light frequencies to control biochemical circuits using photo-electronic circuits. Life has used photonics for 3.8 billion years. Humans have been toying with this process for 6-8 thousand years. The remnants of those hacks can be seen in monuments man left scattered across the globe. Photonics can overcome the high-speed electronic interconnect bottlenecks common in electronic circuits. Direct optical connections between processing elements provided many advantages to Mother Nature in building living systems. These include low signal loss, minimal power requirements, high efficiency, and data transfer over fiberoptic cables that provide massive connects to all parts of the system with minimal energy or information losses. Is there an example we can use in a cell to make the point?
Single tubulin proteins, for example, follow precise rules of chemistry and physics to spontaneously self-assemble, or polymerize, into the microtubules essential for cell transport and motility. The proteins’ binding interactions effect rules that specify how the pieces fit together to form the resulting structure. They also specify when and how tubulins assemble from a nucleation complex—a molecular algorithm governing the logic of polymerization. Mother Nature created an culture of connectiveness in tissues linking to the colony of mitochondria in our tissues. No cell is untouched in photonic circuitry. Tubulins form microtubules in the brain. Inside of them water is found confinded at small scales. Water confined at 30 nm reacts very differently than water does at 1.4 nm. Life wants the water a mitochondria makes confined at small scale. This is another reason this water has little deuterium present in it. At one end of the tubulins the openings are larger and at the other end the spaces is smaller by design. This is designed to confine water to harness its optical abilities when it becomes a special liquid crystalline structure where the quantum thermodynamics is allowed to occur.
These complex structures self-assemble with remarkably few mistakes. Though considered quite simple, little is understood about the principles that govern programmable structural order underlying this type of spontaneous self-assembly. We now have clues it does not require energy to occur. Darwinian evolution should no longer be a special state or case of matter, but thought of as a more general phenomenon of how biophysical processes naturally organize matter using the operational laws of quantum mechanics on a planet.
The local star of that planet provides most or all of the power, but the thermodynamic state of the planet set the floor and ceiling of what kind and type of living systems can be built. The fundamental principles that govern how macroscopic properties emerge from microscopic interactions and arrangements really should be the cornerstones of biologic sciences. Today it is not because we are ignore the hidden variables in nature. It appears the inherent entropy in the system is useful to self assembly. Microtubules are just another example of aperiodic crystals themselves. This mimics the picture I showed you of my rhubarb earlier in the series.
In crystals, the simplest example of spontaneous self-assembly, subunits of the whole are arranged in a repeating pattern that extends indefinitely in all directions. If you know the position of one unit in the pattern, you can tell the exact position of every other unit. Life appears to depends in large part on storing and processing information in this way.
For genetic material to carry the diverse instructions required for living processes, Schroedinger initially proposed, it must be stored in an “aperiodic crystal”. Just nine years later, it was clear that DNA is indeed an aperiodic crystal, when another physicist Francis Crick showed us that genetic information in DNA/RNA is conveyed through this irregular pattern in its matter. Much like computers, biological systems are programmed to follow a precise set of rules, or algorithms, to store information and solve problems. These biological algorithms direct actions biophysically in a myriad of ways to biochemical processes to create complex patterns and structures by chemically modifying and assembling individual components. We have to realize that biophysics and biochemical arms work in entangled fashion. If we do not teach our clinicians about these connections and patients suffer as a result. This is what quantum thermodynamics does. It extends the classical laws of thermodynamics to explain what life really is.
SUMMARY:
The water your mitochondria makes, that surround your proteins is a time crystal. Your entire brain is floating in it. Your mindset and th eability to think is directly related to the crystalline formation possible or impossible that is developed by the water in the CSF that sits on top of your neocortex.That is the crystal that your eye communicates with via electromagnetic pulses it senses via the retina.
All time crystals are a form of matter that “ticks” when exposed to an electromagnetic pulse—from sunlight via our eyes, skin, or gut.
Time crystals, first identified in 2016, are quite different than the crystal above. Their atoms spin periodically, first in one direction and then in another, as a pulsating force is used to flip them. You should remember that mezmorizing gif from QT#6 as an example of how this happens. That’s the “ticking of the clock mechanism.” In addition, the ticking in a time crystal is locked at a particular frequency, a specific resonance even when the pulse flips are imperfect because of the changing diurnal aspects of the sun.
This implies all of biology is applied chemistry from how light interacts with electrons and protons inside of us. Chemistry is applied physics, so every living thing on this planet runs a “resonance software program” built by hydrated proteins. These proteins are conserved magnetically in DNA. These proteins are changed by the addition of electrons, protons, and light. Proteins respond only to specific frequencies of light. They have their own ‘zip code’.
That zip code is more important than the genetic code because it is changes every second of every day we live. Our proteins are surrounded in a sea of water our mitochondria makes from substrates from food and the atmosphere which all become programmed by the sun at our current location. Proteins are quantum Garmin GPS devices fundamentally. The action of water around proteins helps quantize the electrons and protons in us to change the matter that makes us up minute to minute. Our species is constantly changing because of these unseen variations.
Evolution is not some static process that occurs slowly. It is ruthless and rapid. We just do not see or understand how the addition and subtraction of electrons orders and disorders us. Blue light and purple light change how our time crystals resonate. The change in that pattern alters what frequencies we connect to because free radicals are changed. This change, cause our resonance to change instantly. Who we were yesterday is not who we are now. That is how fast evolution is operating in us. Darwin’s version of the process leaves much of that detail out.
What we once use to respond to at one point in our life, we remain limp too, as the light we surround ourselves changes. What once was an abyss becomes a new echo chamber that dominates our life. This is how electromagnetic pollution is reprogramming our us constantly. This is how people develop tinnitus and electrohypersensitivity. This quantized process occurs by the varying kinetic energy buried in frequencies of sunlight. What happens if we bury the sun and build neon God’s? This entropy causes the melody of life to change. The beats our hearts and brains used to march to can be changed by light when we get right down to it. Light is an electromagnetic wave that gets changed to an electro-mechanical vibration by proteins and water around the protein-capture the sound blast for signaling. Changing your light environment changes your inner terrain and the dis-ease train will not be able to thrive at your station of life. That is what “zip code” is to a mitochondriac.
What on the surface seemed ludicrous when I began this blog now suggests entropy drives order. This now opens more questions for biology and medicine. You’ve got to question beliefs to understand nature’ innovation using the physics we know is possible. Just because something is unlikely does not mean it cannot happen. This is why I reject Occam’s razor arguments in science. What nature does not forbid will happen given enough time for the environment to sculpt the change using non-coding DNA, light, water, and mtDNA.
Photons of light come to Earth and to humans in a timeless state. Electrons and protons in proteins in living systems pay attention and resonate with that light to absorb the energy and information light contains. Our cells then transmute that into other waves, light and sound, which start our internal clock ticking to create time from light. The next time you listen to a human heart beat, feel a pulse, or listen to breath, you are sensing someone else’s zip code. There is information in those waves. You will be drawn to them or recoil from them. This is how you connect with other living systems to build your network.
Here is a present for my Patrons and members. I told you you would all get the benefits of reading snipets from my future works if you remain a Patron. This is the “early” realease of my own biohack from 2003-2005 of my own EHS reversal from my Operating room lights and Wifi.
I have told this story to many of my patients with EHS.
After hearing it, many have made the tough decision to relocate, mitigate and rebuild their redox potential with sunlight using information quanta mechanisms that nature provides cells to relearn using light. It sound bizarre until you understand how sunlight passes information from quantum spin to orbital angular momentum properly. (Stay tuned for QT #7) Many do not do enough and they remain on their way to getting severe EHS as their immune cells lose massive amounts of information quanta from the AC power grid.
I’ve been asked my opinion if EHS can begin as a transient mild headache while in the proximity of a 50Hz or 60Hz Magnetic field (fan or transformer) and progresses as the bodies learn from the information quanta in the man-made fields of the electric power grid via the mechanism in the QT series on Patreon? The answer is yes that is precisly how EHS goes from bad stimulus to learned behavior. This can manifest as a sign of transient mild PTC by destructuration of HDW in the coherent domains of the EZ. It can cause changes or inflammation of the pia of the brain where our main SQUID is located.
In 2003 I found I was getting tuned to the things in my OR when we went to papers EMR’s. The number one offender for me was LED lights and the Wifi from anesthesia machines. I found in my hacks after they showed up in my work environment my sleep was degraded quickly so I went searching. I began concentrating transition metals because of the OR EMF and blue light the hospital added to the OR. So I started to change my OR start times and I went outside between each case to get the sun to teach my system with solar EMF’s. Therefore I knew I had to use CT when I got home in the sun with a higher fat diet to get my redox potential back in my inner mitochondrial membrane to lose weight and reduce the EHS symptoms causing my cognitive haze. I realized I had to get my ATPase spinning faster without using excess food (ECT flows) to do it. This is when I realize this is what UV-A light does for cytochrome c oxidase via its release of nitric oxide from the skin and arterioles in the skin that come to the surface when UV-a light is sensed. I also realized the”pressure wave it made was the key to making the soliton in the blood to cause the NO release.
The addition of the sun and fat increased my ability to change the water made in my matrix that was attuned to the OR nnEMF’s. I found doing this also to supercharged my P450 system via the PPP. This was an unexpected result of the hack.
This is how I learned to always choose redox over detox. The PPP gives us back the major reducing element, NADPH. I realized the H in NADPH had to be light hydrogen to properly make intracellular glutathione in cells. This is why I no longer will use the new version of IV tylenol that hospitals are psuhing for patients in surgery. I have found it reduces glutathione the most of any drug in our surgical toolbox. In fact, if one uses oral tylenol often you should drink DDW to reduce the chance of getting EHS in an altered magnetic field. The NADPH acts to recycle glutathione and the glutathione links directly to the melatonin cycle in the brain. It is also when I found out that saturated fat diets and DDW increases endogenous glutathione production. Then I found evidence for this in the literature.
That is when my life really changed its focus. When glutathione is low, we must add back UVA light to increase apoptosis and this help raises melatonin at the same time. Often EHS destroys melatonin cycles in the brain with the Vitamin A cycle. All opsins use Vitamin A to work with light. This is how I found the LED lights in my OR were making me sick. It is also when I made the prediction that melanopsin would be found in the skin. This is why skin problems develop in our patients before they get really sick from electro pollution. It is also why my profession uses Vitamin A, in retin A, to try to improve these issues. Is the redox is bad it makes on worse? If the redox is decent it can help. The smarter move for EHS people is getting AM sun when the UVA IRA transition occurs. It dawned on me then that that artificial light was fully capable of altering people to make them more electrosensitive and this change would lead to poor cogntive function that made it harder for them to think, learn, and adapt. It was why I was losing my edge in surgery for something I truly liked to do. It all occured because of the light hospitals adapted too to save money in the 2000’s.
This is how I figured out the blue light transition metal link too in 2004, as well. Once I fixed my redox problem using the sun, I was able to fix two problems for the price of one. Deep CT I found invaluable for my transition metal issue. It was the only thing that helped my ASI and melatonin because of the chronic OR exposure I faced. It is also when I realized I would have to get out of the OR at night. That is why my actions are different than others. I think you get the biology……but the irony is that this all caused me to look at my own lifestyle modifications as a physicist would……lifestyle changes are really a superposition of the quantum state…….once you embrace a new probability it can and does happen in your reality This is how information quanta changed my own life in 2005.
It is also why I do things very differently in the operating room now as a result. I had to keep my ability to think paramount if I was going to get well.
Water puts the “magnetic” back in “electromagnetic.” In nature’s schematic above, a standing light wave with magnetic (blue) and electric (red) field peaks is generated in an optical photonic cavity, it acts as a resonator. As the light bounces between two reflection points that act like mirrors it transfers information from the quantum spin of particles to orbital angular momentum. This is how lasers can be tuned and how we can increase the base quantum spin number of photons. Normally photons have a quantum spin number of one.
Most people have no idea that light appears to be able to have unlimited storage capacity for OAM when we use an optical resonator. What does this physically in us? Water does this. Not the water from your tap, or the water you drink. It is done by the water your mitochondrial makes. I will be talking about this process in three weeks in Vermont at Shelburne Farms. you might want to get some tickets. You can review my talk on photosynthesis from 2016 and my talk on the retina from 2017 on Youtube. This year there won’t be a YouTube video so attendance will be helpful.
Once water is physically changed by sunlight it obtains new physical properties that give it unlimited nonlinear potential. Researchers have now proven this to be true in several sub-disciplines of physics. How much do you know about aquaphotomics? If you do not, you might want to join my tribe of misfits at https://jackkruse.com/become-a-member/
If you cannot swing that join me here at Patreon. For in-depth blogs on the coming age of quantum, biology becomes a Patron and I will teach you and answer your questions directly for the cost of a coffee a month. https://www.patreon.com/DrJackKruse
ARE YOU READY TO BECOME A BLACK SWAN MITOCHONDRIAC?