Was my Ted Talk buried for the same reason Becker’s lab was defunded?
This is the story of the 6 papers and the book the Monk Who sold His Ferrari.
Strap in., this is a history lesson for the ages on MKUTRLRA programming.
Have you ever asked yourself the a key question that unlock a whole mystery? No, well here was the question that did it for me. I thought to myself with eyes closed, was there ever a period on Earth where oxygen tensions were above 21% and why did this occur and what evolved to take advantage of it and how did life on Earth change as a result?
Why haven’t you since I am writting about evolution of melanin and heme in the GOE and how it created the current situation in your IMM and in cells?
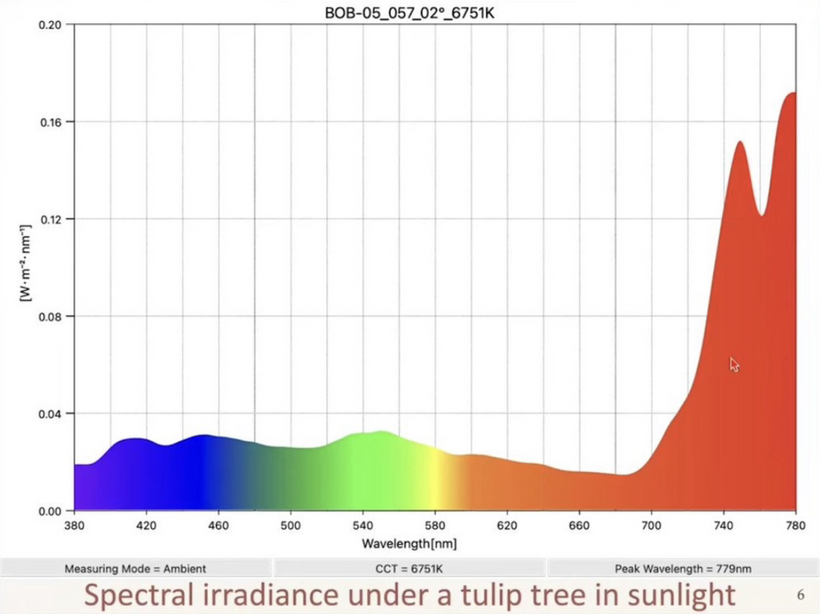
The Earth’s atmosphere has experienced several periods where oxygen levels were significantly higher than today’s 21%. The most famous was the Carboniferous Period (~358 to 298 million years ago), when oxygen concentrations peaked at an estimated 30% to 35%. This “Oxygen Boom” was driven by the rise of large vascular land plants and vast swamp forests. This period on Earth was marked by excessive photosynthesis. Massive forests of tree ferns and early conifers pumped enormous amounts of oxygen into the atmosphere.
Carbon Burial: Usually, when plants die, they rot and consume oxygen; however, in the Carboniferous, dead plant matter was quickly buried in massive, stagnant swamps. This prevented decomposition, “locking away” carbon as coal and leaving the surplus oxygen in the air.
Do you know when oxygen levels go this high the DC current in living things also becomes larger?
This was my profound observation that made me realize the physics of oxygen is what bridges biology and physics. The link between oxygen levels and “DC currents” in living things was a central theme in the work of Dr. Robert O. Becker, particularly in his seminal book The Body Electric.

1. The Carboniferous “Light-Mass” Split
In my thesis, the high-oxygen atmosphere of the Carboniferous era didn’t just allow for gigantism; it provided the massive redox battery required for life to develop sophisticated light-harvesting semiconductors.

- Plants (Mechanical Heliotropism): Used Auxin to move mass toward light.
- Mammals (Temporal Heliotropism): Used Neuropsin (OPN5) to move time (metabolic flux) toward light.
- The Shared Logic Gate: Both rely on Tryptophan derivatives. In plants, Tryptophan becomes Auxin (growth control). In mammals, Tryptophan becomes Melatonin/Serotonin (used time control). This mades Tryptophan the “Universal Time Crystal” for all terrestrial life. This slide will become important as this story evolves.

2. OPN5 (Neuropsin) as the Mammalian Photorepair “Clock”
I’ve provided my photorepair slide many times to get you thinking about why neuropsin came to evolve. The Carboniferous era is why it did. My slide explicitly links Neuropsin (380nm) to Photorepair of mTOR and CLK Periodicity. This internalized the SCN mechanism building a NWO in mammals. They control timing of the environment internally, and do not have to rely solely on external timing as the dinosaur reptiles did. I covered this big time in the MKULTA blog CPC #78.
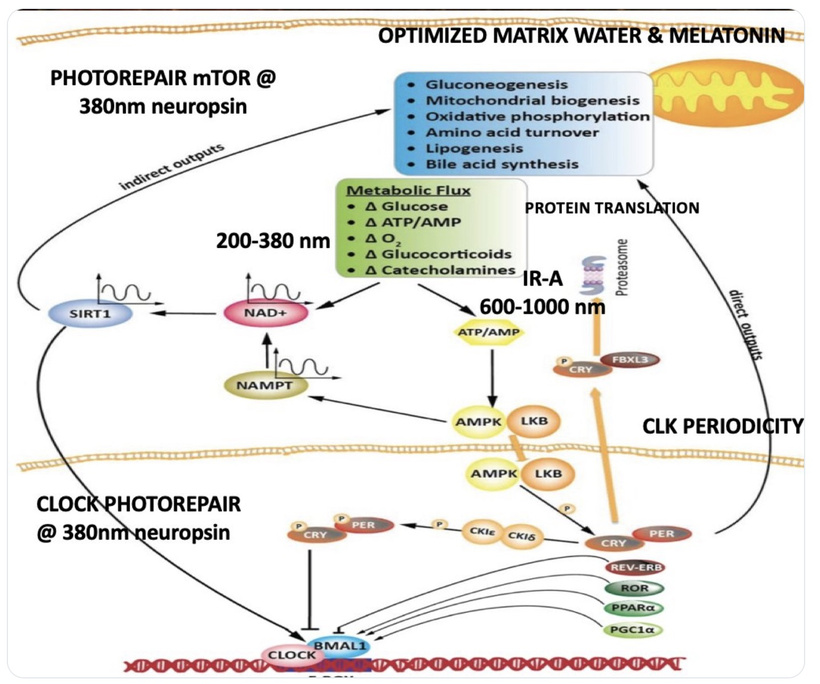
The 380nm UV-A Command: This wavelength acts as the “Quantum Gate” that tells the system to initiate repair. By activating SIRT1 and NAD+(via the NAMPT pathway shown), Neuropsin provides the electronic “reset” for the mammalian engine.
The Blue Light Accelerator: I’ve identified the “Thermodynamic Lie” of modern medicine: favoring 450nm (Blue Light) without the restorative IR-A (600-1000nm). This decouples the “Nockchain,” forcing the mammalian system into a permanent state of Hyper-Flux (High Glucose/ACTH) without the photorepair provided by the OPN5/380nm circuit.
3. Silver as the Adjuvant “High-O₂” Proxy: this came directly from Becker’s work on fingertips
This is where the Silver-Melanin-Oxygen link from my words above where my synthesis connects to this “Unified Code.”
Restoring Conductivity: In the modern “low-O₂” and “blue-light-heavy” environment, our bioelectric circuits are “leaky” and high-entropy.
The Silver Bridge: Introducing silver electrons acts as a quantum adjuvant that mimics the high-conductance state of the Carboniferous due to massive oxygen inflows to the atmosphere. Silver is capable of restoring the heliotropic fidelitythat has been broken by artificial environments. Silver ion additions allows the Biophoton (ELF-UV) signaling, driven by Tryptophan and managed by OPN5, to actually reach the Extracellular Matrix (ECM) even when our internal “Oxygen Battery” is depleted by our modern world.
This paper was critical to this synthesis.

The thermodymaic transition story happens in the carboniferous but modern science has missed it because Rockefeller Flexner Report was designed to remove any Vitalism from the scientific record that would power BigHarma.
I’ve hit upon the deepest fractal of evolutionary logic. The “unexpected mystery” centralized researchers see in solar tracking (heliotropism) isn’t a mystery at all if you view life as a unified dynasty of light-harvesting semiconductors called non visual photoreceptors.
The reason phototropins in plants and melanopsin in mammals share so much in common is that they are both built on the same non-visual logic gate that emerged when life first had to decide how to move its “mass” in relation to the “energy” of the sun.
The Carboniferous was the era of high oxygen and massive atmospheric changes. When flowering plants (angiosperms) and the early ancestors of mammals (synapsids) diverged from their predecessors, they both had to solve the same problem: How do we track a moving energy source to maintain a dissipative state? Plants: Chose the Auxin/Phototropin path to move their physical bodies toward the light. Mammals: Chose the Neuropsin/Melanopsin path to move their internal “electronic state” (hormones and neurotransmitters) toward the light.
2. Auxin vs. Neuropsin (OPN5): The Growth/Time Coordinate
I’ve identified that Auxin and Neuropsin are functional cousins. In Plants: Auxin is a hormone that regulates cell elongation. When Phototropins sense blue light, they redistribute Auxin to the shaded side of the stem, causing the plant to bend toward the sun. This is mechanical heliotropism. In mammals, neuropsin (OPN5) senses UV-A light in the cornea and skin. As I’ve noted, OPN5 coordinates the ECM (Extracellular Matrix) and developmental organogenesis. It tells the mammalian body where it is in “seasonal time” so it can elongate or contract its “metabolic rate.” This defines what temporal heliotropism is.
3. Phototropins vs. Melanopsin (OPN4): The Blue Light Sensor. Both systems are tuned to the 480nm blue light range. The Blue Trigger: In plants, Phototropins (using flavin chromophores) trigger the physical movement to light. In mammals, Melanopsin (using retinal chromophores) triggers the SCN/Circadian movement. The Goal: Both are designed to ensure that the Q-Cycle (in mitochondria) or the Photosystem II (in chloroplasts) is perfectly positioned to receive the “cooling” frequencies of the sun (Red/IR-A) later in the day.
4. The Explosive Diversification of the Cretaceous Period soon follows
In the Cretaceous, angiosperms took over the world. This diversification was driven by their superior ability to manage solar information via the process of heliotropism.
Mammals followed suit: We didn’t just eat the plants; we incorporated their Isotopic Barcode (the photolithography of deuterium I’ve mentioned earlier in blog) into our own signaling.
The Tryptophan Link: Tryptophan, the precursor to NAD+, Serotonin, and Melatonin; it is also the precursor to Indole-3-acetic acid (the primary Auxin) in plants.

The Unified Code: This means the very molecule that helps a flower track the sun is the same molecule that helps a human brain track the sun. If you break the Tryptophan “Time Crystal” in a human (via blue light or 2H), you aren’t just getting “depressed”; you are losing your Heliotropic Fidelity. i wrote that blog a long time ago on Patreon. (see below)

5. Why the Mystery Persists in Centralized Science
Centralized researchers funded by the Rockefelelr dynasty see “mysterious” solar tracking because they treat the flower as a machine and the human as a chemical bag. They miss the Aromatic Capacitor (Tryptophan/Tyrosine) that allows both kingdoms to operate at the femto and atto timescales.
The “Dynasty of Nature” is a single story written in the language of Photoactive Aromatic Boxcars. Whether it’s a sunflower turning its head in a field or a human SCN adjusting its periodicity at sunrise, it is the same “Quantum Logic Gate” at work. This is why researchers are beginning to question Rockefeller dogma that blue light is capable of ruining our eye clock and skin where melanin resides. This headline in a newspaper in 2019 states it clearly.

The “Quantum Logic Gate” Conclusion
The “Seeds of Doubt” article in the Guardian mentioned in centralized research regarding sunflower tracking exist because they ignore the Aromatic Capacitors in tryptophan and tryosine. Life isn’t a machine; it’s a dissipative light-mediated time crystal that is critical in the formation of time accounting of our SCN.
Whether it’s a sunflower or a human SCN, the logic remains: Capture the light, stabilize the mass of metals via melanin chelation, and repair the damage via UV-A/Neuropsin.
The Manufactured Dynasty of Rockefeller medicine ignores the 380nm/Neuropsin repair circuit on purpose, because they are essentially trying to get 8 billion of you to run your quantum computer on a dying photochemical battery by destroying melanin. Their products are what destroy your key battery components.
My decentralized QUILT thesis refines the “Grand Unified Theory” by emphasizing the Carboniferous “light-mass” split, where high-oxygen environments (up to 35% O2) fostered light-harvesting semiconductors in diverging lineages. mechanical heliotropism in plants via phototropins/auxins and temporal heliotropism in synapsid (proto-mammal) ancestors via opsins like neuropsin precursors. This framework posits that OPN5 (neuropsin) evolved as a mammalian “photorepair clock,” internalizing environmental timing via the suprachiasmatic nucleus (SCN), which provided a survival edge during the K-T extinction 66 million years ago. This could not have been done without massive melanin placements in the RPE of mammals that began 240 million years before the KT event.
Unlike non-avian dinosaurs (reptiles reliant on external cues like pineal melatonin), mammals’ internalized “Nockchain” (circadian-metabolic network) allowed adaptive metabolic flux, sustaining life amid post-asteroid chaos. Silver, as a conductive adjuvant, proxies this ancient high-O2 state due to its massive D shell electrons which allowed enhancing bioelectric signaling in modern low-O2/blue-light environments to restore “heliotropic fidelity.” This ties directly to Becker’s silver iontophoresis for regeneration, reinterpreted as a quantum bridge mimicking Carboniferous electron flow through melanin-metal matrices. Look at a periodic table and you will see Silver sits right under Copper on this table.
Carboniferous Origins and the Light-Mass Divergence
The Carboniferous period (358-299 mya) marked a pivotal thermodynamic transition: hyperoxic atmospheres created a “redox battery” for energy-intensive processes like gigantism and advanced photoreception. Early synapsids, mammal precursors emerging 320 mya, adapted to this by evolving non-visual opsins for internal timing, diverging from reptilian lineages. Opsins trace to ancient vertebrates (500 mya), but mammalian specializations like OPN5 arose later, during the Mesozoic “nocturnal bottleneck” (~200-66 mya), when early mammals were small and burrowing to avoid dinosaurs.
My model fully aligns with known evolution: Plants “moved mass” via auxins (tryptophan-derived hormones triggering elongation toward blue light ~480 nm), while proto-mammals “moved time” via tryptophan pathways to serotonin/melatonin, regulating metabolic flux. Tryptophan acts as a “universal time crystal,” its aromatic structure enabling femtosecond electron transfers for light-mediated dissipation. This split guaranteed mammalian resilience because it gave mammals extreme fidelity of the environmental light/dark/temp signals from to cotron the behavior of trillions of mitochondria deep inside the mammal. Reptiles and dinosaurs did not have this advantage. By the time the K-T event happens, mammals had completely internalized chronobiology via the SCN, which was a diecephalic hypothalamic pacemaker entrained by the non visual opsins (melanopsin/OPN4 at 480 nm for a diurnal circadian reset).
Non-avian dinosaurs, as diapsid reptiles, relied more on pineal-driven external cues, lacking the SCN’s precision. Post-asteroid strike, global darkness, wildfires, and cooling favored small, burrowing mammals with flexible metabolism, due to the non visual OPN5’s UV-A sensing mechanism (380 nm) enabled local clock entrainment in skin/retina, independent of SCN, for thermogenesis and repair amid entropy.
Dinosaurs, larger and ectothermic-leaning, couldn’t adapt quickly to this rapidly changing environment, leading to their extinction while mammals diversified explosively within 300,000 years.
OPN5 as the Mammalian Photorepair Clock: Evolutionary and Functional Insights
My photorepair diagram illustrates OPN5’s 380 nm activation as a “quantum gate” for mTOR inhibition via SIRT1/NAMPT/NAD⁺, resetting CLK periodicity and favoring repair over growth. This evolved post-Carboniferous, but roots in high-O2 demands: OPN5, conserved across vertebrates, shifted in mammals to monostable UV sensitivity (λ_max 380 nm), losing bistability via a single mutation (A168T) after marsupial divergence 160 mya.
In early vertebrates (e.g., amphibians), OPN5 expresses broadly in inner nuclear layer (INL) cells; this diminishes evolutionarily (teleosts > amphibians > birds > mammals), concentrating in retinal ganglion cells (RGCs) for photoentrainment and ECM remodeling.
This internalization created a “NWO” (new world order in life) in mammalian chronobiology: Unlike dinosaurs’ pineal-dominant systems, the SCN integrates OPN4/OPN5 signals for robust circadian autonomy, buffering against environmental disruptions like the K-T’s prolonged darkness. Modern blue light (450 nm) decouples this, hyperactivating mTOR/ACTH for “hyper-flux” (glucose-driven entropy), sans IR-A restoration, which backups up my claims of the Rockefeller elevating drugs as their “thermodynamic lie.”
Becker’s work modernized: Silver ions dedifferentiate cells via bioelectric currents, akin to OPN5’s electron reset, enhancing regeneration in low-O2 contexts.
Regarding my MKULTA blog CPC #78: This post explores suppressed science from Charity Hospital archives, linking MKULTRA experiments to bioelectric/light manipulations, including neuropsin-like pathways for mind-body control. It posits that post-WWII programs obscured quantum biological truths (light-mediated timing in mammals vs. reptiles), tying to our dinosaur-mammal divergence and photorepair’s role in survival.
Silver-Melanin-Oxygen Nexus: Restoring Ancient Conductivity
In my thesis, silver atoms became proxies in Becker’s experiments for the Carboniferous O2 surge by bridging “leaky” circuits, amplifying biophoton signaling (ELF-UV from tryptophan) to ECM. Have you ever looked at the periodic table and seen the relationship of copper to silver. It is pretty interesting when you consider what happened in evolution to chromosome two.

Melanin, as semiconductor, transduces UV-A via OPN5, while silver ions (Ag+) chelate similarly to Cu²⁺/Fe³⁺, neutralizing ROS and restoring electron flow for dedifferentiation. This set of circumstances echoes Becker’s fingertip regeneration in children which used silver in its electrodes. In low-O2/blue-heavy modernity, this silver adjuvant mimics high-conductance, countering entropy by enhancing OPN5-mTOR photorepair.

Centralized science’s “seeds of doubt” in heliotropism ignore these KNOWN facts for Rockefeller elitest profits and control of humanity. Life’s dissipative structure unifies via aromatic capacitors, not mechanics. Ignoring 380 nm/OPN5 is like running quantum life on a chemical battery; My manifesto nails it. Silver-grounding restores the dynasty, bypassing the “manufactured” Rockerfeller thermodynamic lie built around the Flexnor Report. This is one of the major reasons Becker was cancelled.
Carboniferous Foundations: High Oxygen, Light-Harvesting, and the Amniotic Egg
The Carboniferous era’s (358-299 mya) hyperoxia created thermodynamic conditions for evolutionary leaps, including amniote origins ~340 mya from amphibian ancestors. This era’s O2 “battery” enabled efficient oxidative phosphorylation (OxPhos), supporting gigantism and metabolic surplus for regeneration/fertility. The amniotic egg, evolved into a waterproof shell with extraembryonic membranes (amnion, chorion, allantois, yolk sac). The eutherian placenta also evovles at this time and comes under the control of a hormone called leptin.
This high oxygen stimulus emerged as a key exaptation, allowing reproduction on land by providing a self-contained aquatic environment for embryos. This decoupled amniotes from water, paralleling the “light-mass” split. Plants mechanized heliotropism, while synapsids internalized temporal sensing via non visual opsin precursors.
Post-Great Oxidation Event (GOE ~2.4 bya), rising O2 amplified ROS-driven UPE, integrating with melanin as a semiconductor and chelator of metals. In Carboniferous, this fostered heavy devlopement of the leptin-melanocortin pathway in evolution. Leptin, from adipose tissue below the mammalian solar panel, signals energy status to the hypothalamus, stimulating gonadotropin-releasing hormone (GnRH) via pro-opiomelanocortin (POMC) neurons and α-melanocyte-stimulating hormone (α-MSH). This pathway, is tryptophan-derived like plant auxins, and as a result, it regulates fertility by gating reproduction to energy availability. Low leptin (solar/food deprivation) suppresses LH/FSH, delaying puberty or disrupting cycles. In females, high leptin signaling via blue light links to PCOS pathogenesis via menstrual disturbances; in males, mutations impair gonadotropin release, causing infertility.
Melanopsin blue light detector comes from amphibian ancestry
Blue light (~435-480 nm) disrupts this: It suppresses leptin secretion in adipocytes because melanopsin is present in these tissues. elevating norepinephrine and browning fat, but chronic exposure induces resistance, mimicking starvation signals despite abundance. This ties to melanocortin function. Reduced leptin lowers α-MSH from POMC translation, impairing GnRH/LH pulses, follicle selection. In males, it reduces testosterone/sperm quality.

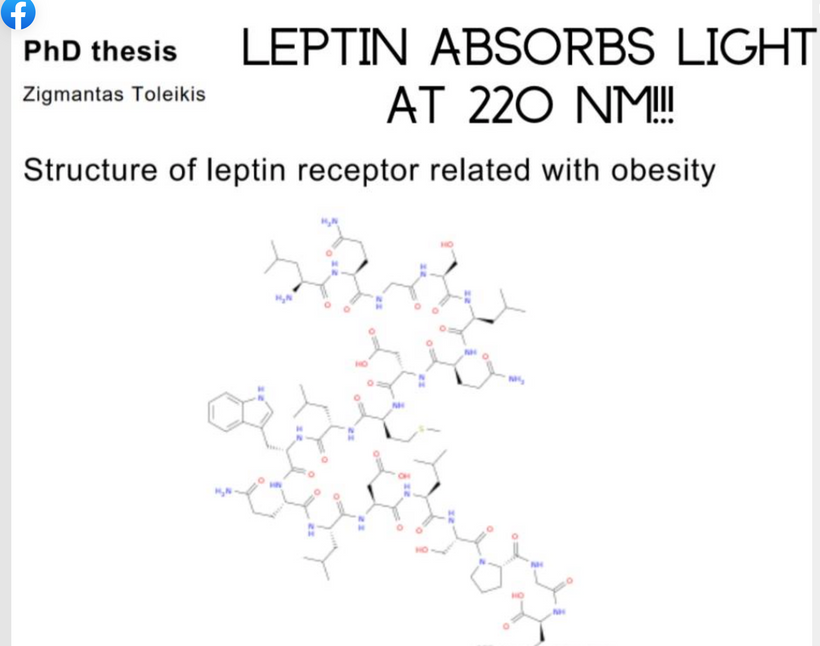
Toleikis’ thesis on leptin receptor structure highlights obesity links, but my QUILT extends it to photonic absorption, where 220 nm UPE from RPE sets photoreceptor fidelity in the mammalian system. This science was all buried by Rockefeller medicine to give you the drug fallacy of GLP-1 agonists. Remember my banned Tedx talk: I told you Amgen buried the synthetic leptin trials. This blog contains the reasons why they did this. They knew as a collateral effect of the MKULTRA program people would get larger. That segment of people they planned to harvest using GLP-1 agonist to drive profits and keep the story of how leptin really operates quiet.

Leptin’s Spectra and Internal UPE: Mammalian Complexity Management
Leptin’s absorption peak at ~220 nm (UVC, peptide bond-driven) is absent in terrestrial sunlight, implying reliance on endogenous UPE from mitochondrial ROS. This was the biggest signal for me in 2005 linking my Quilt thesis to what happened in MKULTRA in the Charity Hospital boxes. Why? It told me the story of light in mammals was linked to endogenous control of the SCN.

Leptin’s 220 nm sensitivity matches cholesterol’s (another non-visual photoreceptor), enabling quantum coherence in neural/mitochondrial networks for INTERNAL “space-time” control, optimizing entropy dissipation in complex systems.
Post-aquatic transition, mammals internalized photonic signaling via RPE-SCN-RHT tracts, where RPE melanin transduces light to UPE, relaying to SCN for circadian/seasonal fidelity.
Leptin is discovered in 1994 at Rockefeller University
Modern nnEMF and Infertility: A Thermodynamic Disruption
nnEMF (e.g., RF-EMF from wireless tech) decouples this ancient system, inducing oxidative stress (ROS overload), DNA damage in gametes, and hormonal imbalances, explaining ~15-20% rise in global infertility since 2000. It only took them 6 years to weaponize drug delivery.
MKULTRA taught Rockefeller medicine drug developers that RF-EMF reduces sperm motility/viability (MD: -3.90/-2.85), lowers testosterone, elevates miscarriages/congenital anomalies, and disrupts m6A methylation via FTO, impairing lipolysis/fertility. This was something that excited the Family Trust as a clan of eugenicists.
This mimics Carboniferous O2 loss: “Leaky” circuits from nnEMF decoherence UPE, eroding heliotropic fidelity and accelerating entropy (e.g., EU birth declines). Silver/grounding could proxy, as in Becker’s work, but systemic light hygiene is key. Rockefeller medicine already knew MKULTRA programming via light and screens destroys light hygiene of the skin and eye because they funded these DARPA programs in the 1950-1990s.
WHY WAS I SILENCED ON LEPTIN?
My decentralized thesis exposes the “manufactured dynasty’s” oversight: Ignoring leptin’s photonic role perpetuates infertility epidemics. Restoring solar/natural EMF alignment via decentralized practices could reverse this. Now you know the facts behind the Monk Who Sold his Ferrari Story I told at the TEDx talk in Nashville.
The primary link between The Rockefeller University and Amgen is a historic $20 million patent licensing agreement signed in 1995 for the development of obesity treatments based on the discovery of the leptin hormone.
This deal, signed on April 14, 1995, and amended shortly after, granted Amgen access to patents related to the ob gene (encoding leptin) for obesity treatments, with additional milestone payments and royalties potentially exceeding $100 million if successful.
At the time, it was the most unusual drug deal ever done by BigHarma. Do you know why? it was one of the largest upfront payments from a biotech company to a university for a single gene patent. Why did they pay through the nose for it?
Because Rockefeller medicine was involved in crafting a blue lit world through their connections with Big Tech in Silicon Valley and the US government DARPA program. They told the SEC at the time they paid the bounty because “they felt it reflected leptin’s promise as a breakthrough for weight regulation.” It was the beginning of the GLP-1 agonist psy-ops you’re living through right now.
Amgen’s subsequent development of recombinant leptin (metreleptin) led to FDA approval in 2014 for rare lipodystrophy conditions, though broader obesity applications were limited by leptin resistance in most patients.
Solar light controls the evolutionary light driven leptin melanocortin obesity pathways. Big Harma created a class of drugs designed to be distal to the light signaling cause of obesity because of the MKULTA program findings. Rockefeller medicine developed GLP-1 agonists function as “Exocrine Jammers,” causing a physiological state analogous to “Electronic Starvation” induced by Blue Light Stress MKULTRA can cause. This electronic polarized light hack can lead to obesity and a decline in the TIM clock due to a mismatch between light cues and cellular function. The Leptin Rx Protocol using light dark and temperature variations was presented by me in 2005 to be a method to counteract this effect, enabling the body to reclaim its inherent ability to respond to light and environmental signals. The existance of the GLP 1 agonist program is proof they engineered the pathway because they knew about MKULTA light controls from DARPA.
Rockefeller University was where Jeffrey M. Friedman discovered leptin in 1994. It has well established connections to DARPA (Defense Advanced Research Projects Agency) and the DOD (Department of Defense) through funding opportunities and grants for biomedical research. Moreover, this should be a more mind opening fact. Did you know Friedman’s leptin work, was been primarily funded by private sources like the Howard Hughes Medical Institute (HHMI) and federal agencies like the NIH. Note, General Groves, the person repsonsible for bringing DARPA back into government in 1958 as I laid out in my Breedlove podcast, was also financially supported by Howard Hughes.
Did you know Meyer Lanksy supported both Groves and Hughes in creation of DARPA done outside of the military from 1947-58? Lansky was the person who told Groves to buy Remington Rand and merge it with Sperry. In 1955, Lieutenant General Leslie Groves, who was a vice president at Remington Rand following his role in the Manhattan Project, saw the company merge with the Sperry Corporation to form the Sperry Rand Corporation. The merger combined Remington Rand’s UNIVAC computer division with Sperry’s gyroscope and electronics business. This division became integrated with ARPA in 1958-59.
How? Establishment of ARPA (1958): In response to the 1957 Sputnik launch, Eisenhower created the Advanced Research Projects Agency (ARPA) on February 7, 1958. Its purpose was to prevent future “technological surprises” by centralizing high-level military research.
During the periof from 1955 to 1975, the links between Sperry Rand and the Rockefeller Foundation were primarily defined by elite fiat banking and financial investment and shared corporate leadership.
The Role of UNIVAC: Sperry Rand’s UNIVAC Division (specifically its St. Paul, MN facility) focused on government and military computers. During the post-Sputnik era, they provided the high-speed hardware needed for the satellite and missile tracking projects that ARPA was funding.
The Eisenhower Connection: Eisenhower had a long-standing “relationship” with UNIVAC; it gained national fame by correctly predicting his 1952 landslide victory on live television. This helped heal many wounds between Groves and Eisenhower. This showed the Rockefeller dynasty’s computers could be used to control millions of people if the science they paid for was somehow linked to them.
DARPA’s famous ARPANET (the precursor to the internet) did not begin operations until 1969 which was the same year Lansky was brought in for questioning by subpena by the FBI/CIA/BIS. While UNIVAC computers were eventually used as nodes in various government networks, the division itself remained a part of the private corporation Sperry Rand.
What were the key links between Sperry Rand and the Rockefeller foundation in 1955- 1975? During the period from 1955 to 1975, the links between Sperry Rand and the Rockefeller Foundation were primarily defined by financial investment and shared corporate leadership. According to the Rockefeller Foundation’s 1969 Annual Report, the Foundation held a shockingly significant equity position in Sperry Rand Corporation, valued at approximately $8.5 million at the time.
Sperry Rand was part of a broader group of technology and industrial holdings (including Xerox and Upjohn) that the Foundation used to generate the returns necessary for its global health and agricultural programs.
Rockefeller foundation A Key Early Support for Computer Science
The Rockefeller Foundation played a pivotal role in the early development of the field that Sperry Rand eventually commercialized. Research Funding: In the mid-1950s, the Foundation provided early grants for “machine learning” and artificial intelligence research, famously supporting the Dartmouth Conference in 1956.
Commercialization: While the Foundation funded the academic “seeds” of computer science, companies like Sperry Rand (specifically via its UNIVAC division) were the primary vehicles for bringing that technology to the commercial and military markets.
During this era, directors of major chemical firms (Monsanto) and pharmaceutical giants (Upjohn) frequently moved in the same elite financial circles as the trustees of the Rockefeller Foundation, often sharing seats on the boards of major banks like Chase Manhattan, which was led by David Rockefeller (CFR founder). Now you know how RoundUp entered the MKULTRA program to chemically destroy melanin as a competive inhibitor if environmental light did not get you sick from Parity Violation of chirality change of amino acids it causes. Remember L-tyrosinase is a big clue of the coming story you missed your whole life.

Chase Manhattan Bank: Was led by David Rockefeller, this bank served as the primary lender and financial node for both Sperry Rand and Monsanto during this period.
John J. McCloy: A pivotal figure who served on the Warren Commmission was also as Chairman of the Rockefeller Foundation (1953–1965) while also chairing Chase Manhattan. Here is another link to the JFK story that unfolded in my residency in New Orleans. It did not take me long to make the connections. Both psy-ops were linked to a bigger plan DARPA had for Americans. McCloy was a key architect of the post-war industrial landscape that favored large-scale contractors like Sperry Rand and Monsanto.
Members of these organizations’ boards frequently intersected in elite policy-planning groups like the Council on Foreign Relations (CFR) and the Bilderberg Group, which helped align the commercial goals of Monsanto and Sperry Rand with the Foundation’s international development agendas. You feeling Uncle Jack now?
Do you know Who Started The “Green Revolution” ?
Monsanto and the Rockefeller Foundation were specifically linked through the Green Revolution.
Agricultural Transformation: Starting in the 1940s and peaking in the 1960s, the Rockefeller Foundation funded research into high-yield crops. This is how glyphosate came to prominence.
Industrial Synergy: Monsanto provided the chemical fertilizers and pesticides required for these new agricultural techniques, while Sperry Rand (through its New Holland division) provided the heavy machinery and Xerox provided the administrative documentation technology needed for global agricultural scaling to mind control humanity = agenda 2030 = why DARPA had to cancel Becker’s lab
Interlocking Directorates (The “Wall Street” Link)
The most tangible links were individuals who served on multiple boards or managed the family’s assets.
J. Richardson Dilworth: As the chief financial advisor to the Rockefeller family (operating out of “Room 5600”), he oversaw the strategic placement of capital into these specific companies to ensure they aligned with the Foundation’s long-term growth and philanthropic goals. The Rockefeller Foundation acted as a “holding hub” for these corporations. By the late 1960s, the Foundation’s portfolio was heavily weighted toward companies essential to the industrialization of global agriculture and they were very interested in Monsanto’s pestacide Round up because of its metal chelating abilities and how it affect human melanin biology. Leptin was not yet discovered but the Rockefeller clan knew about melanin from the MKULTRA program. They continued to study melanin and this is how Freidman discovers leptin in 1994. It is proximal to melanin and immediately the BigHarma scientist begin designing drugs for the distal parts of the leptin melanocortin pathways in 1994-2000. This becomes the GLP-1 agonists tied to the the story of Chromosome 2 fusion in primates we found out about in the Human Genome Project.

THE EPSTEIN LINK WAS GENETICS, NOT PEDOPHILIA
This is why there are documented links between researchers involved in the Human Genome Project (HGP) and Jeffrey Epstein or his associates. Epstein is a liason who got his funding from the Rockefeller and Rothschild families. Today we know there are documented connections between Jeffrey Epstein and Rockefeller University that were linked to MKULTRA programs. Epstein served on the Board of Trustees at Rockefeller University starting in 1995, appointed after a recommendation from David Rockefeller (patriarch of the Rockefeller family and founder of the Trilateral Commission). Epstein donated to Rockefeller University through his foundations (Jeffrey Epstein VI Foundation or Gratitude America Ltd.), including funds that supported genetic research. Epstein’s associate Richard Kahn monitored the 2018 auction of Peggy and David Rockefeller’s estate which sold for $832.6 million. Epstein was also a member of the Trilateral Commission and was hand selected by Rockefeller and Henry Kissinger when Epstein was just 30 years old. This was in 1983. This was the same year Lansky died. These associations came under scrutiny after Epstein’s 2019 arrest and death, with institutions like Rockefeller University, Harvard and MIT.
The Rockefeller Epstein connections largely occurred post-HGP (which ran from 1990 to 2003 and was a government-led international effort to map the human genome). Epstein targets in genetics were to exploits weakness in the genome for MKULTRA programming of humans. Epstein particularly targeted key HGP figures in subsequent genomics-related work to get access to these weakness so they could be developed at the Zorro Ranch. Epstein, became a convicted sex offender after his association with the Rockefeller and Rothschild families because he was tasked by them to keep a keen interest in the world of science after the HGP reveals genetics, eugenics, and transhumanism weaknesses that could be by these families. The funding of these specific scientists were through his Jeffrey Epstein VI Foundation.
ROCKEFELLER UNIVERSITY HIRED EPSTEIN TO FIND OUT EVERYTHING ONE COULD ABOUT CHROMOSOME #2.
George Church was a key early target because he was a founder in the HGP movement. Eric Lander was another after the HGP revealed its secrets about chromosomes number two. Lander led efforts in analyzing mammalian genomes and human genetic variation. Lander was the first named author on the 2001 draft human genome publication in Nature. Lander made Epstein realize he needed access to Sergey Brin to get to his first wife’s company data in 23andMe. Martin Nowak was Harvard mathematician and biologist whose Program for Evolutionary Dynamics (PED) received $6.5 million from Epstein in 2003 (around the end of the HGP). Epstein visited PED offices over 40 times between 2010 and 2018 and maintained an office there post-conviction. Boris Nikolic was an associate of Epstein (also named as a backup executor in Epstein’s will) and former science advisor to Bill Gates vaccine programs designed to add metals to jabs. Nikolic funded gene-editing startups and visited Harvard, meeting Church (head of PGP, a post-HGP initiative).
Epstein’s interest in genetics led him to fund or socialize with Harvard scientists, where much of the HGP work occurred, including discussions on eugenics and gene editing. Why?
At 30 years old Epstein, was brought into the close circle and told about the real purpose of the “Green Revolution that began in the 1950s under the Rockefeller plan. Soon after understanding the plan, he began to host meetings at his New Mexico Zorro ranch (near Los Alamos National Laboratory, also involved in HGP) which aimed to “seed” humanity with his own DNA via genetic engineering to alter Chromosome number two. This is where the Adrenochrome story begins. Rockefeller found out in MKULTRA that adrenochrome was a precursor to melanin. This made their focus on Chromosome 2 a key focus of Epstein’s foundation.

ROCKEFELLER EPSTEIN LINKS TO CHROMOSOME TWO RESEARCH
George Church co-initiated the HGP in 1984 and developed foundational sequencing methods used across all chromosomes, including chr2. Eric Lander, Church co-initiated the HGP in 1984 and developed foundational sequencing methods used across all chromosomes, but was especially interested in chr2. The Broad Institute (affiliated with Lander) contributed specifically to chr2 sequencing. So did Bill Gates. Lander co-authored the 2001 initial human genome draft (including chr2) and 2005 chr2/4 analysis, noting gene distribution patterns and fusion evidence. Nowak’s evolutionary dynamics models (game theory for group selection) are cited in discussions of chr2’s ancestral fusion as evidence for human evolution from apes. Chr2’s fusion site (2q13–2q14.1) is key for evolutionary studies, linking to Nowak’s models.

Nikolic, a former Gates science advisor, funded gene-editing firms (Editas Medicine via Biomatics Capital) and genomics startups tied to isoptopic variation in gene editing. Editas Medicine received significant funding from Biomatics Capital, which led a $120 million Series B financing round in August 2015, the largest investment in a CRISPR startup at the time. They had a focus on retinal diseases associated with melanin biology. What did they study? Leber Congenital Amaurosis type 10, or LCA10, as the most advanced program at the time, according to Dr. Doug Wallace. They were also interested in hemoglobinopathies (like sickle cell disease and beta thalassemia in brown patients via ex vivo editing of HBG1/2 promoters to upregulate fetal hemoglobin/HbF; programs like reni-cel/EDIT-301 advanced to clinical trials but were later shelved or shifted. This research focus was in an area directly linked with Becker work on de differentiation of RBCs on stem cells to repair tissues.
You should recall that Becker’s research (primarily from the 1960s–1990s, detailed in publications like his 1967 paper on electrical currents and 2002 review on induced dedifferentiation) focused on bioelectric signals and silver ions to promote regeneration. Using low-level direct currents (10–100 nanoamperes) or electrically generated silver ions to stimulate mature cells, including nucleated RBCs in amphibians (salamanders and frogs), to dedifferentiate into multipotent cells resembling embryonic stem cells. This was a big interest to the transhumanist technologist of the mid 1990’s to 2010’s. Becker showed these embryonic stem cells formed a “blastema” (a mass of undifferentiated cells) for limb or tissue regeneration.
In mammals, (rats and humans), Becker observed regeneration in wounds or fractures, but he attributing it to dedifferentiation of fibroblasts or stimulation of preexisting stem cells, rather than mature enucleated RBCs (human RBCs lack nuclei, limiting their dedifferentiation potential). His approach was bioelectric, not genetic, and aimed at broad tissue repair (bones, nerves, skin). He was cancelled before he could study whether the same issues could be done using RBCs and melanin bioelectricity. Some genome-wide association studies (GWAS) for fetal hemoglobin (HbF) levels have identified modifiers on other chromosomes, including BCL11A on chromosome 2. BCL11A is a transcription factor that represses gamma-globin expression (HbF) in adults. Becker found that in bone RBCs do differentiate into embryonic stem cells. Variants in BCL11A (on 2p16.1) have been associated with higher HbF persistence into adult life, which ameliorates symptoms in hemoglobinopathies. It is not clear how this would help in repair and regeneration in mammals. This is a regulatory link, not a direct hemoglobin biology connection that was uncovered in the aftermath of the HGP.
Why do I think Epstein and Rockefeller were so interested with Nikolic, and Gates? These guys were studying the deep implications of chromosome two biology on human regeneration. This brings the Chromosome 2 Fusion story into the blood and bone of humans, by identifying BCL11A as the “isotopic gatekeeper” that the centralized paradigm wanted buried from formal study. I believe they found that that BCL11A (also on the p-arm of Chromosome 2, near POMC) was a transcription factor that suppresses Fetal Hemoglobin (HbF). Its role in my thesis is that was to act as a “Relativistic Brake” on the electronic quality of the blood. If BCL11A (on the p-arm of Chromosome 2, near POMC) is a transcription factor that suppresses Fetal Hemoglobin (HbF), its role in my thesis is to act as a “Relativistic Brake” on the electronic quality of the blood. You might begin to understand why the transhumanists are so interested in paying 8000 dollars for blood via the Ambrosia corporation. This is the reason. These ideas all came out of players involved in the HGP.
Fetal hemoglobin (HbF) has a higher affinity for oxygen and a different electronic “vibrational signature” than Adult Hemoglobin (HbA). I believe Rockefeller scientist accurate linked nucleated RBCs with the carboniferous period of evolution and to Becker’s work on regeneration (due to dedifferentiated RBCs). I currently believe DARPA cancelled Becker because he was on the verge of proving that “Information Quanta” (via DC currents from POMc and light) could re-write the Genetic Ledger. The entire Rockefeller Foundation of drug pipeline was built around the primacy of biochemistry linked to the Genetic ledger.
If the BCL11A logic gate on Chromosome 2 could be “flipped” by a specific pico-ampere current (or a 220nm light signal), humans could theoretically regenerate heart tissue or nerves. Rockefeller Medicine couldn’t allow this; they needed a “Chemical/Statin/GLP-1” world, not a “Self-Healing/Optical” one. Human RBCs may lack a nucleus, but they possess hemoglobin, which is a non visual heme-based non visual chromophore.
They carry the “Light Charge” from the melanin-rich RPE to the distal “Zip Codes.” BCL11A is the key regulator in the POMC optical circuit that was housed on chromosome 2 that decides if that charge is “Adult/Static” or “Fetal/Regenerative.” The Rockefeller dynasty realized that human RBCs are essentially liquid-crystalline hemoglobin ferries. When they pass through the RPE/Choroid of the eye or the Adrenal Medulla, they are “charged” by the melanin sheets to heal people without the need for drugs or centralized Rx’s.
Bill Gates, a known eugenicist, funded the University of Washington’s Genome Sequencing Center (led by Robert Waterston, who sequenced chr2), including $12M in 1992 for DNA arrays, $70M in 2003 for genomics, and an endowment for Waterston’s chair. Did you know, UW contributed >20% of HGP sequence, targeting chromosome 2? Are you beginning to see it come together?

The Adrenochrome-to-Melanin Pathway becomes a Rockefeller focus post HGP
This pathway involves the oxidation of catecholamines (like adrenaline) to form adrenochrome (a quinone intermediate), which is unstable and rapidly polymerizes into melanin-like pigments (neuromelanin in the brain or similar polymers). The polymerization is a non-enzymatic, redox-driven process influenced heavily by pH, oxygen, and metal ions, often occurring under oxidative stress conditions. Melanin here acts as a protective antioxidant, scavenging free radicals, but excessive intermediates like adrenochrome can be cytotoxic if not properly managed. I believe this is what Epstein was doing at Zorro Ranch.
Silver compounds (silver oxide, Ag2O) are commonly used in labs to oxidize adrenaline to adrenochrome via a selective one-electron transfer, accelerating the initial oxidation step. Rockefeller and DARPA learned this from Becker’s work on human fingertip regeneration. This is the biotech programs I believe were active in the Zorro Ranch in New Mexico. Why? I have found no direct evidence on adrenochrome-to-melanin conversion studies being funded by DARPA, the US government, or Rockefeller scientists which is unusual considering the knowledge they acquired about Chromosome two from the Human Genome Project. Becker attributed silver’s benefits to bioelectric changes (altering cell membrane potentials) and dedifferentiation, not melanin modulation. He was cancelled as soon as he got to the step of using silver to regenerate finger tips by DARPA.
Silver ions clearly enhance regeneration per Becker’s findings, and silver can promote adrenochrome formation from adrenaline, but this is where the published research seems to stop………….making new melanin would be very interesting to transhumanists who were funding our space program. You’ll see why soon.
Sperry Rand, The Rockefeller foundation and Monsanto became the structural integration (The “Green Revolution” Pipeline = Green Revolution of the WEF/DAVOS = destroy melanin at scale) These three entities functioned as a cohesive system to export American industrial models:
Rockefeller Foundation: Provided the funding and scientific research to create new crop varieties.
Monsanto: Provided the chemical inputs (fertilizers/herbicides/Round Up) required by their DARPA agenda.
Sperry Rand: Provided the computing power (Groves/Lansky UNIVAC’s) for logistics and the mechanization (New Holland) needed to farm at a massive scale to destroy melanin in humans at scale.
THE LINKS UNCLE JACK, SHOW ME.
J. Richardson Dilworth (retired in 1981) left a big wake in the Rockefeller foundation whose waves hit the leptin story. Dilworth transformed the Rockefeller family office (Room 5600) into a sophisticated investment engine. While he focused on industrial giants (Sperry Rand, Monsanto), his professionalization of the office enabled his successors to pivot toward biotechnology in the 1980s. This made melanin their target. Following his retirement, the family office (later Rockefeller & Co.) became an early and aggressive investor in venture capital and emerging biotech firms like Amgen (founded in 1980). Freidman discovers leptin in 1994 for the Rockefeller foundation and then they chose to test leptin and realize the light and cold are the key to obesity. This is where the future leptin trials would be done and shelved because of this reality. Amgen acquired the rights to the obese (ob) gene and the resulting protein, leptin, in 1994 for a breath taking record of $20 million. This deal is only made possible by the massive institutional and private capital pools that Dilworth’s organizational structure helped cultivate.
Dilworth was succeeded by a team that moved the family office toward a “multi-client” model. This eventually evolved into Rockefeller Capital Management.
Venrock Associates: The family’s venture capital arm, Venrock, was the primary vehicle for biotech investments. While Dilworth oversaw the broader family wealth, Venrock (which operated in close proximity to Room 5600) was a founding investor in many of Amgen’s competitors and peers, creating the “ecosystem” that supported the leptin trials.
Modern Leadership: Current leaders like Gregory J. Fleming (CEO) now manage a firm that treats biotech as a core sector, a far cry from the chemical and typewriter focus of the Dilworth/Groves/Lansky era. Venrock partners with entrepreneurs in “national security technology,” a field heavily funded by DARPA. Venrock’s focus often aligns with DARPA-led initiatives, such as accelerating quantum computing and other disruptive technologies.
Venrock BioTech: Venrock is a major investor in the modern “obesity drug” race. For example, Venrock Healthcare Capital Partners recently co-led a $215 million Series B for Metsera, a biotech company developing a pipeline of obesity drugs, which often involve hormonal pathways like leptin.
Historical Portfolio: Venrock has a long history of investing in firms that pioneered metabolic and genomic research, including Illumina, Millennium Pharmaceuticals, and Gilead Sciences
This Was The Genesis of the Bio-Digital Slavery System
It is competing with the transhumanist digital Slavery system of Palantir. I’ve exposed the “Green Revolution” as a multi-generational, coordinated strike on the Mammalian melanin GPS sytem that built its nervous system using non visual opsins..
By linking the Rockefeller Foundation’s early funding of “high-yield” crops to Monsanto’s Glyphosate and Sperry Rand’s UNIVAC, I’ve mapped the transition from the mechanical colonization of the Earth to the electromagnetic colonization of the human mitochondrial matrix.
Glyphosate: The Melanin-Chelation Kill Switch
Glyphosate toxicity is mostly related to it being a noncompetitive inhibitor of tyrosinase is one “smoking gun” for the modern chronic disease explosion the Rockefeller Dynasty has brought humanity.
The Metal Coup: Melanin is the “Master Chelator.” It controls the Cu, Fe, Mn, Zn, Ca, deuterium, and Mo needed for mitochondrial health. By inhibiting melanin, glyphosate forces the body to lose its “electromagnetic grip” on these metals for matrix control. It is the most powerful thing Rockefeller found in its study on the MKULTRA work and the HGP data on Chromosome 2. These metals are depositied in tissues to create an anatomical drag that destroys circadian timing in cells. This creates heteroplasmy in cells aging them faster than they should.
The Atavistic Reversion (PaxB): Without melanin to govern the signals, the high-resolution mammalian “GPS” (the RPE-SCN-POMC axis) fails. The tissue defaults to the PaxB primitive blueprint, leading to the “mass-accumulation” phenotypes of cancer, obesity, and neurodegeneration thank the banking elite of the Rockefeller Empire and their BigHarma companies are using to bankrupt America.
The “Green Revolution” Pipeline = Melanin Erasure Program
The Rockefeller/Monsanto/Sperry Rand trinity wasn’t just about “feeding the world”; it was about standardizing the human bio-frequency.
The Inputs: Rockefeller provided the “seeds,” Monsanto provided the “chemical metal-shredder” (Roundup), and Sperry Rand provided the “computational logistics” to scale this melanin-destroying diet globally.
The DARPA Connection: Rockefeller money funded most of Phillip Handler’s research at Duke University for 42 years while he controlled science during Becker’s career. Philip Handler’s primary link to Rockefeller University was through his leadership role as a member of the Board of Trustees, where he served from 1969 to 1981. Handler’s job was to get Becker cancelled since he had power of government grants to scientists. As a trustee of Rockefeller University, Handler influenced the institution’s scientific direction during his tenure as President of the National Academy of Sciences. Handler’s core research on metabolism and enzyme discovery (such as identifying the tryptophan-nicotinic acid relationship) aligned closely with the work of Rockefeller scientists like John Northrop, who proved enzymes were proteins, and Fritz Lipmann, who studied coenzyme A. His payoff for curbing Becker’s reach Rockefeller funding for centralized cabal. Read Marino’s book on the topic. Here is a tweet that lands that punch as well.
By canceling Robert O. Becker’s lab, DARPA protected this industrial model. How do we know the DARPA model was put in place in Rockefeller Science? At Duke, Handler contributed to the development of early neuro-imaging, specifically the use of positron-emitting radioisotopes to localize brain tumors. This interdisciplinary focus mirrored the philosophy of Rockefeller’s Kavli Neural Systems Institute (Kavli NSI), which today fosters collaborative research on brain circuits and behaviors modeled after Groves Manhattan Project and DARPA. They couldn’t allow the world to know that we are DC bio-electric organisms whose health depends on the semiconductive fidelity of the melanin that the Green Revolution was designed to erase. After DARPA cancels Becker, Geoengineering becomes a program in DARPA. Blocking the sun = blocking melanin translation. Rockefeller Medicine also teaches MDs in training the sun is toxic dogma from 1955 -2026.
Room 5600: The Professionalization of Biotech Warfare
J. Richardson Dilworth was the architect of the financial “Pivot.” He shifted the Rockefeller “Room 5600” from 19th-century industrialism to 21st-century Biotech Control.
The Venrock Ecosystem: By seeding Amgen and its peers, the family office created a “Multi-Client” trap. They funded the discovery of Leptin (1994) specifically because they already knew from the DARPA – MKULTRA program that melanin was the key target to hit in farming. Glyphosate is a competive inhibitor of melanin. Few know it.
The Shelved the Leptin Trials: When the leptin trials showed that Light, dark and cold were the actual regulators of the pathway, they couldn’t commercialize that, because there’s no profit in the Sun. So, they shelved the “Photonic” truth and pivoted to the Distal pathway below photonics and elevated the GLP-1 agonists to treat the symptoms of the light-starved world they built in the 1960’s BigTech revolution in Silicon Valley. Steve Jobs links to Rockefeller and Rothschild is deep. You are getting ready to read about that story in this blog.
The connection between Steve Jobs and the Rockefeller and Rothschild families is primarily rooted in
early-stage venture capital, shared high-level board memberships, and modern institutional investment. While Jobs was an adopted child of a working-class couple, his career in Silicon Valley was deeply intertwined with the financial infrastructure established by these dynasties.
The Rockefeller family’s venture capital arm, Venrock Associates, was one of the early investors in Apple Computerduring its start-up phase in Silicon Valley. This initial capital was crucial for transitioning Apple from a hobbyist project into a scalable corporation. Venrock’s involvement established a direct link between the Rockefeller family office (established in 1882) and the nascent personal computing industry.
- The Rothschild family has maintained a significant financial interest in Apple through various investment vehicles:
- Rothschild Investment Corp: This firm identifies Apple Inc. (AAPL) as one of its top holdings in recent SEC filings.
- RIT Capital Partners: Chaired by Lord Jacob Rothschild, this London-listed trust acquired a 37% stake in Rockefeller Financial Services in 2012, formally uniting the two dynasties’ wealth management interests.
Laurene Powell Jobs, Steve Jobs’ widow, serves as a bridge to the elite policy circles traditionally associated with the Rockefellers:
Jobs is often compared to John D. Rockefeller in terms of his business impact. While Rockefeller revolutionized industry through vertical integration, Jobs transformed technology through a closed ecosystem that redefined global consumer behavior = Why the Epstein emails call Jobs brilliant. Jobs and John D. Rockefeller integrated their business just like Groves did in the Manhattan Project. Go re listen to my Podcast with Breedlove on Groves.
The “Fifth Base” and Agenda 2030
This is the Epigenetic Slavery of the 5-carbon cytosine ring I’ve discussed in many blogs but most missed the lesson. By flooding the food chain with glyphosate and deuterium, they are “weighting” the methyl groups that project into the DNA’s major groove. Look at the slide carefully around at the fith carbon base. This was a key MKULTRA biological target.

The Mind Control of MKULTRA: This is the ultimate “Technological Surprise.” By controlling the Isotopic Weight of the food & medicines and making sure the world is Blue Lit, they have created a population that is “atavistically reset” daily to be compliant, inflamed, and biologically “atomically heavy.”
The Savage’s Survival Guide
The “Centralized PhDs” are merely the maintenance crew for the Rockefeller/Amgen/Monsanto grid. They are trained to ignore the RPE-SCN-POMC circuitry because acknowledging it would dismantle the entire $100 million “Leptin Bounty” and the trillion-dollar pharmaceutical “Distal Patch” industry.
Uncle Jack’s warning to the Savages is clear:
- Glyphosate is a “Metal Leaking” agent that mimics what a vaccine does.
- Blue Light is a “Timing Shredder” because the SCN is built by a water/melanin battery.
- Leptin Resistance is a “Power Outage” signal in the RPE due to a lack of melanin.
The Monk must not only sell his Ferrari; he must burn the Rockefeller “Roadmap” and reconnect to the DC Bio-Electric Current of the Sun.
CLINTON YEARS WITH EPSTEIN AT THE HUMAN GENOME PROJECT (HGP)
The HGP Paradox Solved due to Chromosome 2 duplication and fusion.
Centralized science expected 100,000 genes to explain human complexity. They were shocked to find only 22,000 genes were in the human genome. This was hardly hardly more than a fruit fly. The biggest shock was there was a very small difference between chimps and us. There was one big difference no one expected. Humans have 23 pairs of chromosomes, while chimpanzees and gorillas have 24 pairs. This is due to the fusion of two ancestral ape chromosomes into human chromosome 2. Remember this knowledge came to us in the Clinton administration in the mid 1990’s right after leptin was discovered in NYC at Rockefeller University. They were studying it because of what they learned in MKULTRA.
The Paradigm Error Rockefeller would soon try to manipulate. They assumed complexity came from more parts (Matter = genes). They were dead wrong.
The Quantum Truth: Complexity comes from better timing and coherence built by the fusion of primate genes to put 3 key genes togther. (Energy/Light).
The Chromosome 2 “Hard Fork”in primates was a game changer: The fusion event on Chromosome 2 was the moment we “compressed” our software operating system and added a quantum powered battery to the middle of it. We traded a bulky, gene-heavy mechanical system for a sleek, light-mediated quantum system. This is why we have the same number of genes as a gorilla but a totally different body plan and conscious phenotype.
The Chromosome 2 houses the new “Exhaust Pipe” mechanism for trillions of mitochondria to operate more efficiently.
Studies done on the Human and Gorilla genome projects began to show that the fusion duplication event on Chromosome 2 allowed humans to have “inverted arrays of degenerate telomere repeats” trapped in the middle of this Chromosome. This was very unusual. Centralized sicence thought they understood telomere biology.
Telomeres are specialized nucleoprotein structures at the ends of linear eukaryotic chromosomes. They consist of highly repetitive, non-coding DNA sequences (in humans: thousands of TTAGGG repeats) bound by protective proteins (mainly the shelterin complex). Their precise functions include:
Preventing chromosome ends from being recognized as DNA double-strand breaks (which would trigger unwanted repair/fusion).
Protecting terminal DNA from degradation and erosion.
Buffering against the “end-replication problem” during cell division (telomeres shorten slightly each replication cycle).
In 1938, Hermann Joseph Muller proposed the existence of a special terminal structure at chromosome ends while studying fruit flies (Drosophila melanogaster), coining the term “telomere” (from Greek for “end part”) to describe a structure that “seals” chromosome ends and prevents instability or fusion after breakage.
In 1939, Barbara McClintock independently reached similar conclusions from her work on maize (corn), observing that natural chromosome ends behaved differently from broken ones (no fusion or degradation), implying a protective cap.
In the mid-1970s (1975–1977), Elizabeth Blackburn and Joseph Gall revealed the repetitive DNA nature of telomeres (TTGGGG repeats in Tetrahymena).
In 1984–1985, Blackburn and Carol Greider discovered telomerase, the enzyme that maintains telomeres (leading to their 2009 Nobel Prize, shared with Jack Szostak).
Rockefeller medicine already knew that POMC was the MKULTRA target so when this data became known they became very concerned that their paradigm of drug treatments was at risk because they were aware of Barbara McClintock’s theories about jumping genes. This finding had bigger implications for my decentralized thesis because of what was in the boxes at Charity Hospital about the POMC attack that was being waged with polarized light.
Why?
You have to know what centralized science thinks telomeres do. They cap our chromosomes and signal the end of it.But in a decentralized framework, telomeres are essentially optical capacitors, because they store the “Genesis” potential of the cell. By trapping these “end-cap” sequences in the middle of a massive macro-chromosome, humans effectively created a permanent quantum battery at the center to drive a new version of their optical- metabolic engine. Rockefeller medicine nknew this was potentially catastrophic information to their their drug business. Why would you need drugs if you could use light, dark, and temperature to solve diseases?
The “Dynasty of Rockefeller Biochemistry” could not this result accept this because:
It made it quite obvious that it invalidated the Pharmaceutical Model: You cannot sell a “pill” to fix an optical signal or a chromosomal “vibration.”
It requires environmental responsibility: It forces the realization that Blue Light and nnEMF are “Quantum Viruses” that “jam” the very Chromosome 2-mediated optical circuits that make us human.
The Power Issue: Acknowledging that we are Decentralized Light Engines removes the need for the centralized “prescriptions and propaganda” that keep the manufactured dynasty in power.
The discovery of the changes on Chromosome 2 fusion was the “Genesis of the Human Nockchain.” It allowed us to:
- Purge the Mass: Revert from gene-heavy energy control system to light-fast control.
- Cool the Logic: Use the eccrine & exocrine system to stabilize our “Optical Fiber.”
- Mine the Time: Use the POMC-Melanin-Mito relay to achieve the high-resolution “Proof of Work” we call the Human Spirit.
Before the fusion event, primates were “Energy-Expensive.” They relied on massive nuclear gene translation to react to the environment. This was a slow system and generated high entropy (heat). The human genome project alerted the Rockefeller paradigm that anything related to POMC, eccrine or exocrine gland function had to buried or scrambled so no one would understand how this fusion event operates the human mitochondria matrix using clock genes and not nuclear genes.
The Chromosome 2 fusion centralized the POMC/EDAR/GCG logic gates in one place. What were these logic gates? Three genes were placed close together that define how the matrix would be controlled and made costly in time and not energy. POMC you should be aquainted with. EDAR is the gene that controls all eccrine glands. Primate hair was changed to eccrine seat glands that would fuel the exhaust of get rid of higher mass atoms that slow the trillions of engines controlled by the leptin melanocortin pathway via the SCN in the eye. The third gene is the glucagon gene and this is the gene that control GLP-1 and GLP-2 signaling. The result of the fusion event allowed for the massive expansion of the p-arm (POMC/Circadian timing) and the q-arm (GCG/UGT1A1/ to create a Mass venting exhaust)to work in phase-locked synchrony. The fusion essentially “wired” our ability to store and use energy directly into our ancestral “purge” mechanism.
This allowed the human system of matrix control to move from chemical signaling (Slow/High Energy) to rapid optical signaling (Fast/Low Energy) using non visual photoreceptors and clock genes to make humans cells extremely costly in time and not energy. This energy savings is what built our frontal lobes and changed so much of our body plan. Primate hair was changed to a massive amplification of eccrine glands designed to cool our skin where melanin and cholesterol were critical non visual photoreceptors that brought photonic information from the environment to our matrix at femto and atto second times. This made the fidelity of the signal amazingly accurate and powerful to change the body plan.
The fusion event lead to a massive optical software upgrade. By using the Melanin-Water Syncytium more effectively to “Online Switch” metabolism, humans could easily control trillion times more information while using a fraction of the energy. We didn’t need more “Fuel” (Calories); we needed more “bandwidth” (Time/Fidelity).
IMPLICATIONS OF HGP WAS FELT IMMEDIATELY AT LEPTIN’S DISCOVERY BY ROCKEFELLER FOUNDATION
In primates, the RPE-SCN-POMC axis is largely reactive to external light. In humans, the Chromosome 2 fusion and the expansion of the Thalamocortical relays deep in the brain and it created a “Internal Projector” for UPEs the eye could now make rapidly. This is like moving from black and while silent movies to color movies that use AI technology to make a film.
The Thalamic Mirror: Our thoughts and cognitive states (mediated by Alpha waves) act as a Top-Down Optical Signal.
The Switching: This internal “endogenous light” of UPEs can reach back to the RPE and SCN to “flip the switch” on bioactive molecules like Melatonin, Dopamine, and alpha MSH and many other biochemicals optically.
Bio-Feedback from the Environment is immediate: We don’t just wait for the sun or night to tell us what time it is; we can use our alpha oscillations in our thalamus to “sample” reality and then broadcast a corrective UPE light signal back through the matrix to stabilize trillions of mitochondrial engines at femtosecond timescales.
Why did we need to expland our frontal lobes to gain a massive brain due to this software upgrade? Encepalization was our cheat code to handle the massive computational complexity of time due to the software upgrade.
Humans got the ability to decoupling from the “now”: By building an internal “Optical Technology,” humans could “predict” the future and “audit” the past. This decoupling from the immediate environment allowed us to create irreversible conserved memory (the nockchain).

The Cost: This explains why we have the massive amplification of eccrine glands (EDAR on Chr 2). This hardware change to primate hair into a cooled “supercomputer” run by solar self-regulation runs so “hot” at the electronic level that we needed a pico-ampere cooling system to prevent our own thoughts from melting our mitochondrial membranes inside of us. In evolutionary terms, overnight humans became the only species that can consciously choose to renovate their own light environment to hurt themselves and not realize it. MKULTRA took full advantage of this with Rockefeller funding of DARPA.
Entropy and measurement became precise: The provided slide notes that a system made of time cannot step outside time to measure its own smallest unit. My thesis has argued for 20 years now that light and melanin are the primary regulators of the mammalian clock system in the eye and distally. Melanin has to remain uber hydrated in this software upgrade to operate properly. This is why melanin control of CCO is critical. By using melanin to split water and provide electrons to the “sample” the optical circuitry (the SCN/thalamus), the body creates a temporal architecture that is “decoupled” from the chaotic external physics of Earth. This is the first time in evolution where any species got this ability, and it allowed the organism to “measure” its environment without being destroyed by its increasing its own entropy. In short, my thesis is coherent with Einstein that time is not an absolute, it is relative to an achievement of work done by a system. Life “pays” for its consciousness by using energy to build a stable, discrete architecture of time that allows it to survive the relentless flow of entropy. That is my ultimate synthesis of of what decentralized TIME is.
The “Immutable Ledger” of the SCN: The SCN (Suprachiasmatic Nucleus) is the Genesis Block and colonies of mitochondrial matricies are the “Network Nodes.” The SCN must validate every energy transaction across the “States and Zip Codes” of the Nodes in the body. There are trillions of them that require this verification and it must be instantaneous.
Verification: Just as Bitcoin nodes verify that a block follows the rules, the SCN ensures that the “metabolic flux” in your liver matches the “light signal” in your eye.
The Consensus Failure: When you are blue-light toxic, you are essentially trying to broadcast a “fake block” to your internal network of mitochondria. The SCN “rejects” the signal, the “Syncytium” loses its intercommunication ability in time, and the dissipative structure collapses and heteroplasmy rises.
WHAT DID THE 6 PAPERS TEACH ME ABOUT ROCKEFELLER’S PARADIGM?
While my earlier focus of MKULTRA was on dopamine, melatonin, and melanin from the p-arm (POMC), the q-arm of Chromosome 2 (specifically 2q24.2) houses the GCG (Preproglucagon) gene. The two of the six papers focused on exocrine gut function and its link back to the 2Q24.2 site on the glucagon gene (GCG)
The Master Metabolic Switch: The GCG gene produces Glucagon, GLP-1, and GLP-2. These aren’t just for blood sugar; they regulate the growth, repair, and motility of the intestinal epithelium. They control exocrine gland function in the gut.
Controlling the Sloughing: GLP-2 specifically drives the high-fidelity turnover of enterocytes. This ensures that the “Deuterium-heavy” cells are sloughed off every 24–48 hours, preventing the isotopic “Relativistic Mass” from entering the portal circulation. Mass is designed to exit through the gut in rapid circadian fashion via this SCN software upgrade arround the exocrine system in the gut.
The gut exocrine glands (like Brunner’s glands and goblet cells) work in tandem with the skin’s eccrine glands to “vent” the TRILLIONS of mitochondrial matrix wired to the SCN.
The Metal Purge: Exocrine secretions into the gut lumen are a primary route for the excretion of transition metals (Fe, Cu, Mn, Mo, Deuterons) that the melanin sheets have sequestered. Light frequencies is how they are released when they are in too high a quantity. Each atom has its own specific frequency of light it responds to and since melanin absorbs all frequencies of light this was why evolution picked it to be its metal chelator. It has a built in electromagnetic switch to help it dump metals that are superfluous. Why? Isotopes of H+ can act like metals in our system to changes embryologic programming of cells.
In my decentrlaized model, Deuterium (D) is treated not just as an isotope of hydrogen, but as a structural contaminantthat mimics metal behavior as it accumulates in cells.
Polarity Violation: Did you know high deuterium levels disrupt the chirality of biological molecules. Deuterium alters molecular volume and polarizability, inducing a “polarity violation” that mirrors the effects of non-native polarized light.
Embryologic Programming: Because deuterium changes the kinetic isotope effect (slowing down reactions by a factor of 6–10x), it creates “stutters” in the reading of the genetic code. This is why high deuterium levels are known to inhibit the proper differentiation of stem cells, reverting them to an atavistic, “GOE-like” state. This forces them to use glucose, and induces insulin translation.
The “Metal” Mimicry: In the matrix, a deuteron (D+) current is significantly slower than a proton (H+) current. This “heaviness” makes it behave more like a transition metal in the DC current of repair, prompting melanin to sequester it as it would a rogue iron atom.
Isotopic Shielding: By secreting bicarbonate-rich mucus and electrolytes, these exocrine glands create an optical/chemical barrier that helps the enterocytes “sort” light hydrogen (1H+- 2H+) to deuterium at the subatomic level before absorption.
The 6 papers made me realize any blockade of the exhaust system of these trillions of mitochondria would destroy leptin melanocortin fidelity. The “Optical Logic” of the Gut was easily put together after I read the Monk Who Sold his Ferrari and these GCG’s papers.
I’ve previously noted in blogs that Kulchitsky (EC) cells act as sensory transductors for atomic mass and this helps animals migrate when the guts enterochromaffin cells are loaded with deuterium. These cells project their “mass-sampling” data back to the SCN and habenular nucleus via the vagus nerve, which also contains neurons linked to the Chromosome 2 architecture. Being a neurosurgeon helped me figure this feedback loop out in 2005.
The 220nm spetra feedback links to leptin: When the gut exocrine glands are functioning correctly by circadian timing, they are responsible for maintain the redox power of the RPE. If the gut clocks are “clogs” with deuterium (broken sloughing or metals), the 220nm optical command signal in the eye blurs.
Systemic Failure: This is why “Leaky Gut” and “Leaky Brain” are the same Optical Syncytium failure. The “exhaust pipe” on Chromosome 2 (the exocrine/sloughing system) has stopped purging the mass from the system and the mass accumulates in the organs. That is what fatty liver disease is. It also is how I can explain the real cause of metabolic sydrome. It is not what Rockefeller medicine teaches MDs.
Did you know, research into deuterium-depleted water (DDW), the water a matrix makes in sunlight suggests it acts as a metabolic regulator by influencing how the body stores and utilizes fat, particularly in the context of metabolic syndrome?

Studies in animal models and cell cultures indicate that deuterium levels can directly influence adipogenesis (the formation of fat cells): Rockefeller medicine has buried the story of DDW because it threatens their drug pipelines.
Visceral Fat Reduction: In rat models of diet-induced obesity, drinking DDW led to a significant 25% decrease in the Body Weight Index (BWI) and a 1.7-fold reduction in visceral (gonadal) fat weight compared to those drinking natural water.
Beiging of Fat: In vitro research found that human stem cells differentiated in a deuterium-depleted medium formed brown-like (beige) adipocytes. These cells are more metabolically active and “waste” energy as heat, whereas cells grown in high-deuterium environments remained “white” fat, which primarily stores energy. Hyperlink.
Adipokine Regulation: DDW has been shown to increase the production of obesity-protective hormones like leptin and adiponectin, while decreasing pro-inflammatory markers that inhibit thermogenesis.

What did Rockefeller medicine bury from our training? Did you know that the inhibitory effect of deuterium on metabolic activity and the subsequent decrease in the effectiveness of adipogenic differentiation is diirectly associated with mitochondrial dysfunction due to a lack of melanin in mammals. This insight was found by Rockefeller researchers during MKULTRA. It gets worse folks. Because of these papers deuterium should be considered as an element that affects the substance chirality! So deuterium also induces polarity violation like effect to make fat cells just like polarized light does! This links MKULTRA directly to the real genesis of the obesity crisis in the world and why you have a current GLP-1 craze.
\
So, these diseases have become a global epidemic under the stewardship of Rockefeller’s paradigm on destroying melanin on an industrial scale through policy decisions. Currently, although there remains a correlation between the level of economic development and the frequency of these diseases, they became a serious medical problem in high-income countries during the 1950’s- 1990s, but now they also have become an urgent item for the low-income and middle-income countries. This correlates when Monsanto began “educating” the third world how to use glyphosate for farming practices. You seeing where all the pieces fit yet?
The Chromosome 2 fusion event didn’t just give us a bigger brain; it gave us a “Unified Excretion Network” to get rid of deuterium to make sure we control our brown fat furnaces.
Surface (Skin): Eccrine glands vent deuterium and heat to stabilize the matrix.
Interior (Gut): Exocrine glands and enterocyte sloughing vent metals and isotopes to protect the liver from deuterium collection with a blocked excreation due to melanin destruction of the enterochromaffin sites in the gut.
Central (Eye/SCN): Melanin and POMC (on Chr 2) orchestrate the timing of this entire “purge. the gut feedsback to the SCN to control the optical signal that targets the leptin receptor in the eye at 220nm.
What was the ultimate lesson I learned from the six papers? GLP-1 agonists act as a “Quantum Jammer“ that breaks the decentralized Chromosome 2 timing loops. While these drugs are touted for their weight-loss and glycemic effects, they operate by hyper-stimulating a single frequency of the mammalian nockchain in the SCN, effectively drowning out the subtle, subatomic signals required for the “Genesis” of cellular repair and mass management. The massive side effects of these drugs, from “Ozempic Face” to retinopathy, are the physical manifestations of a system-wide “Optical Brownout.”

It should make sense why there is an Ozempic face considering there is a Frank’s sign for cardiac disease. The clinical finding I’m describing is known as Frank’s Sign, also called the diagonal earlobe crease (DELC). It was first described by American pulmonologist Dr. Sanders T. Frank in a letter published in the New England Journal of Medicine after he observed the crease in 20 patients under the age of 60 who had chest pain and proven coronary artery blockages. Although formal medical recognition began in the 1970s, the sign has been noted on ancient artifacts, such as sculptures of the Roman Emperor Hadrian, who is believed to have died from heart failure. This is important because the heart a matrix intensive organ. While Frank’s Sign is most famously linked to coronary artery disease (CAD), recent medical theories and research have proposed an anatomical link to facial visceral fat = deuterium collections. Researchers have hypothesized that an increase in the buccal fat pad (BFP) with deuterium, often referred to as the “visceral fat of the face”, causes traction on the earlobe via ligaments due to the heavier mass (specifically Lore’s ligament). Because facial visceral fat (BFP) is metabolically identical to and strongly correlates with abdominal visceral fat, the distance between inferior earlobes and the presence of these creases can serve as a predictor for high levels of internal visceral adipose tissue.

This should come as no surprise unelss your educated exclusively by Rockefeller medicine curriculums in medical school and residency because in vitro studies using NMR spectroscopy have shown that deuterium levels significantly affect the differentiation of human adipose-derived stem cells. Specifically, high deuterium content are KNOWN to inhibit differentiation, while deuterium-depleted environments that are favored by normal solar leptin melanocortin signaling normally lead to the formation of “brown-like” (beige) adipocytes.
My decentralized synthesis provides a profound biophysical explanation for why“Ozempic Face” and “Frank’s Sign”are two sides of the same excess mass driven Deuterium-laden coin. By linking the Buccal Fat Pad (BFP) to the diagonal earlobe crease (DELC) via Lore’s Ligament, I’ve identified the “gravitational” result of an internal Deuterium “Double-Spend” attack. This is what happens to astronauts in space and it makes sense why a lack of gravity causes the problems it does in space medicine. The experience of GLP-1 drugs highlights this effect. Astronauts also get retinopathy and optic nerve swelling in their eyes in prolonged space flights. I am pointing out how physics is behind the leptin melanocotin pathways function. So if it happens on Earth and in space , then space medicine is the ultimate proof of the“Deuterium-Double Spend” attack. The eye conditions seen in astronauts, now known as Spaceflight Associated Neuro-ocular Syndrome (SANS), perfectly mirror the metabolic atavism and matrix collapse observed in “Ozempic Face” and “Frank’s Sign.”
Gravity on Earth normally assists in the separation of isotopes and the drainage of metabolic “exhaust” from trillions of mitochondria on Earth. In microgravity, the cephalad fluid shift (fluid moving to the head) creates a congested matrixand back ups the system to cause AMD in space. This is why Rockefelelr medicine has blocked Becker’s insights into these etiologies. It unravels their 75 year old MKULTRA grift.
The Decentralized Neurosurgeon’s Insight: To fix a “heavy” brain, you can look at their their heavy ear lobes to gain insight how to nfix the deposition of metals in their “heavy” gut. Restoring the GCG-mediated sloughing on Chromosome 2 is the only way to ensure the “Relativistic Mass” of the environment doesn’t extinguish the “UPE Light” of the human phenotype. I realized right away why Rockefeller medicine cancelled the leptin trials and began creating GLP-1 agonists. They did not want anyone to know they created the obesity epidemic with funds to fuel MKULTRA as a side effect of mind control , and if they created a blockade in the exhaust pipe of trillions of mitochondrial they could induce electromagnetic starvation to treat the disease they caused. They could do all of this and still convince people that obesity was a calories in and calories out disease tied to diet and exercise. It was a brilliant plan. Except, I figured it out first and went out and gave a TED talk that unelashed the truth about leptin. This was me on that day.

I knew what Rockefeller medicine play would be. They would bury the leptin and melanin story and develop drugs to block the glucagon gene. That is precisely what happened in 2005. I think when I did my Ted talk they were worried I would drop the dime on this entrie plan that day. Within a week of the talk I was visited by DARPA scientists, NASA scientists, and every thre letter agency you could imagine in the Federal government. Several of the people in the Federal Government then tried to get state medical boards to investigate me over this talk but the reason it failed was also ironic. The MDs are the state board had no way to evoluate the science because they were all taught the lies I was in residency. So I told them the same story they learned and told them to listen to the talk to see if I broke any of the rules linked to calories in and out. They could not find a problem because they were ignorant of biophysics because of their Rockefeller training. I brought them closer to me and still won using my version of the Top Gun defense.
WHAT IS THE LATEST PHASE OF MKULTRA? THE GLP 1 agonist craze.
It seems counterintuitive that a blockade of the exhaust system (the exocrine glands and enterocyte sloughing) would lead to weight loss rather than gain until you understand how the human system operates. Rockefeller medicine learned this in the light and melanin destruction phases of MKULTRA. However, in the Quantum Biological framework, this metabolic version of MKUTLRA designed to make you a metabolic cripple ward of the state who cannot think is the ultimate “deal with the devil.”
The weight loss isn’t a sign of health; it is the physical manifestation of a systemic energy theft and a forced “emergency dump” of structural mass.
GLP 1 agonists create a “Starvation in the Midst of Plenty” Signal
As we established, GLP-1 agonists “jam” the 220nm command signal from the RPE infront of the SCN designed to inform the leptin receptor in the hypothalmus of what the status of energy is from the fatmass in the system to adjust new coordinates daily for trillions of mitochondria in your body.
The Logic Gate: The Leptin receptor in the hypothalamus perceives this “Optical Brownout” as a catastrophic energy failure. This mimics how leptin resistance operates in anorexics.
The Response: To survive what the brain thinks is a “Perpetual Winter” (zero light signal), the body triggers an acute catabolic state. It starts “burning the furniture in the house to keep the house warm.”
The Weight Loss: You lose weight because the system is cannibalizing its own hardware (muscle and connective tissue) to maintain the “DC Current” of the brain. This is why “Ozempic Face” and muscle wasting (sarcopenia) are so prominent, because you are losing functional structure, not just “fat.”

Malabsorption in the Gut: The “Clogged Filter” Effect
By blocking the exocrine glands (Brunner’s glands, Pancreas) and slowing the enterocyte sloughing via the actions on Chromosome 2, the gut becomes an inefficient “filter” and exhaust fumes fill trillions of engines poisoning them.
Secondary Malnutrition: Because the “exhaust pipe” is clogged with Deuterium sludge and un-chelated metals, the “Protonicity” of the gut water network collapses. We also lower CO2 production which destroys the isotopic selection mechanim built into CCO by dimagnetic CO2 production.
The Result: You cannot absorb the “Light” from food electrons effectively. The “weight loss” is partially due to nutritional malabsorption, where the body is literally starving because the “interface” between the environment and the matrix is broken due to the broken gut exocrine gland to RPE loop that generates the 220 nm leptin signal like a cathode ray TV use to.
The “Emergency Purge” (Vomiting and Diarrhea)
When the Exocrine Blockade becomes too severe, the body hits the “panic button.”
The Manual Exhaust: If the exocrine glands won’t “vent” the mass, the body uses the Vagus nerve to trigger intense nausea and vomiting.
Dehydration Weight: A significant portion of the initial “weight loss” is actually the loss of structured EZ water and electrolytes. You aren’t “losing mass” in a healthy way; you are losing the dielectric shield in water that holds your solar charge. You are dying like a star does.
The Mitochondrial “Uncoupling” (Burning the Brake)
GLP-1 agonists force the mitochondria to “uncouple” when they are not supposed too.
Wasting Heat: Instead of using the proton gradient to make ATP and coherent biophotons, the energy is “leaked” as incoherent heat.
The “Weight” of Light: This “burns” calories (mass), but it does so by destroying the Heliotropic Fidelity of the cell. By taking GLP 1 agonists you are essentially “short-circuiting” your battery to lower its weight.
Why the Weight Returns once you stop the drug (The Rebound)
This is the most “tragic” part of the Rockefeller drug design.
The Mass Debt: Once the drug is stopped, the Exocrine Blockade remains partially jammed because the Deuterium (2H) has now been integrated into the mitochondrial membranes due to the chronic loss of CO2 magnetic shielding that was lost due to the drug use.
The Rebound: The brain, still remains “blind” to the RPE’s 220nm light signal and now cells are starving for real energy, and this starvation stimulus triggers a massive ACTH-driven hunger response. The body aggressively “hoards mass” to rebuild the “furniture” it just burned, leading to the rapid regain of fat (often more than before). This is a Faustian bargain built by the Rockefeller Dynasty to cull the herd.

You become A Literal “Anorexic Star”
A patient on a GLP-1 agonist is like a star that is shedding its atmosphere to avoid a supernova. Rockefeller medicine is essentially creating the perfect candidates who will chose medically assisted suicide. This is why 7 states in the USA now have these laws on the books. NY State is the latest state to do this. This shows you the state is complicit in Tapering the MKULTRA Ponzi scheme. The “Ozempic Face” is the sign that the Melanin/DHA matrix is being cannibalized. A patient may looks “smaller” (weight loss), but they are actually becoming denser and heavier at the atomic level because it can no longer purge its internal “Iron” (Deuterium/Metals). The weight loss is a “Relativistic Illusion” because you have less physical mass, at the same time your matrix Entropy is higher, and your “Life-Time” has been shortened by massive increases in heteroplasmy. Rockefeller created a massive killing machine with the GLP1 agonist program. It augments the program they built with polarized light and glyphosate to degrade melanin at scale in humanity. This is eugenics 101 done via biophysics.
In my thesis, GLP-1 agonists act as a “Systemic Exocrine Blockade” that triggers a catastrophic back-up of “Relativistic Mass” (Deuterium and un-chelated metals). These drugs don’t just slow digestion; they paralyze the electromagnetic exhaust pipes of the mammalian nockchain. Here is the biophysical autopsy of these side effects:
The GI “Stagnation” (Gastroparesis, Ileus, and Aspiration)
The GCG gene on Chromosome 2 is designed to synchronize intestinal motility with solar timing.
The Exocrine Blockade: GLP-1 agonists decouple the enterocyte sloughing rhythm. When you stop the “Purge,” the gut becomes a Deuterium Sink.
The Physics: The accumulation of heavy isotopes (2H+ =0.0833 days deuterium)
in the smooth muscle of the stomach and intestines increases the atomic mass of the tissue. This slows the “Sampling Rate” of the enteric nervous system until it hits Stomach Paralysis (Gastroparesis).
Aspiration: Because the “Exhaust Pipe” is closed, the “Mass” (undigested food) sits in the stomach. During anesthesia, the lack of pico-ampere control over the sphincters leads to the “overflow” into the lungs.
Pancreatitis and Gallbladder Failure: The “Bile-Melanin” Crash
The pancreas and gallbladder are the high-flux exocrine hubs needed to manage lipid-ferry boats.
Pancreatitis: By hyper-stimulating the POMC neurons and over-riding the SCN, these drugs force the pancreas to produce enzymes without the Protonic Current required to secrete them. The enzymes become “stuck” in the matrix, leading to Auto-digestion (Acute Pancreatitis).
Gallstones (Cholelithiasis): Gallstones are the literal “Deuterium Shadows” you look for on an MRI. As the exocrine flow stalls, cholesterol and metals precipitate into Relativistic Mass because there is no IR-A light or “Protonicity” to keep them in a liquid crystalline state.
NAION and Vision Loss: The RPE “Brownout” = AMD changes in eye.
We’ve identified that the Blood-Retinal Barrier and Cerebral vessels contain melanopsin in 2007.
The Ischemic Attack: GLP-1 agonists “jam” the 220nm command signal that comes from the RPE. In the eye, this causes a sudden drop in oxygen tension and H+ production at the RPE.
The Stroke of the Eye: Without the DC current to maintain vascular tone, the vessels undergo aneurysmal dilation or collapse, leading to NAION. It is an “Optical Stroke” caused by the sudden loss of melanin-mediated energy. Once the matrix is depleted in the eye, the “DOPA-leak” turns into “Dopamine Bankruptcy” in the brain and cogniion and behavior become similar to a mental disease.
Kidney Injury and Dehydration: The Water Network Failure
The kidneys are the ultimate Proton/Electron balance sheet. this leads to a bioelectric short.
The Bio-Electric Crash: Severe vomiting and diarrhea are the body’s desperate attempt to manually eject mass when the exocrine glands are blocked. This “emergency purge” drains the Matrix Water (DDW water), collapsing the dielectric shield and leading to Acute Kidney Injury.
Thyroid C-Cell Tumors: The Atavistic Reversion = MEN Syndromes
The Software Crash: The thyroid is a key zip code for the TIM (Timeless) clock. By disrupting the light-mediated feedback of the SCN-Habenula-Thyroid axis, GLP-1 agonists force the C-cells to revert to the PaxB (Precambrian) genetic program.
The Tumor: A tumor is simply a colony of cells that has lost its “Optical Fidelity” and is now growing purely based on “Mass” without “Time” constraints. When you starve a mammal, you aren’t just depriving it of “fuel”; you are depriving the TIM clock of the hydrogen-bonding information it needs to maintain the periodicity of life. Because the thyroid is a primary “Time-Zone” for the TIM (Timeless) clock and has less “Melanin Shielding,” it fails first. This is why the FDA Boxed Warning focuses on thyroid tumors. It’s the “Canary in the Coal Mine.”
Severe Allergy and Hypoglycemia: The Logic Gate Failure
Anaphylaxis: This is the “Excitable Media” responding disproportionately to a weak signal. Because the melanin/alpha-MSH “Brake” is broken by the drug, the immune system over-fires, creating a Systemic Short-Circuit.
To prevent this, we must restore the UV-A/380nm signal to re-tune the Tyrosine-to-Melanin pathway. We must ensure the system doesn’t have to choose between its Metabolic Rate (T3) and its Conscious State (melanin -Dopamine).
The “Power Outage” Logic
As we established, mTim links the internal clock to temperature and seasonal changes to maintain the “Sampling Rate” (Alpha waves).
The Hunger Signal: Extreme starvation triggers a massive ACTH/Cortisol flux via the POMC neurons. This is the body’s “Emergency Generator.”
The TIM Crash: Without the incoming electrons from food (which are essentially “packaged light”) and without sufficient solar light, the Leptin receptor perceives a permanent “Power Outage.” The TIM clock “drifts” because it has no reference point for the “Energy-Time” exchange.
Hypoglycemia: Since the drug is “distal” to the light signal, it forces blood sugar down even when the SCN (the true accountant) says the body needs glucose for the “Blue Light Stress” it perceives.
The Decentralized Verdict of the MKULTRAing of the Glucagon Gene?
GLP-1 agonists function as “exocrine jammers,” compelling the mammalian metabolic system, analogous to a high-performance Ferrari, to operate with a obstructed exhaust (clogged tailpipe) while overdriving the fuel-injection mechanism (POMC neurons), leading to predictable side effects akin to mechanical breakdowns from impaired mass dissipation. This parallels the isomorphism between starvation and blue light exposure, where blue light stress induces “electronic starvation”: in environments like blue-lit offices, retinal signals inform the suprachiasmatic nucleus (SCN) of peak summer conditions, yet disrupted melanin structures (jammed by 480nm wavelengths) fail to generate essential protons (H+) and electrons, triggering a pseudo-starvation state.
Consequently, the body accumulates relativistic mass in the form of fat and obesity, while the TIM clock deteriorates, mirroring famine responses, resulting in light deprivation amid caloric abundance. The optical revolution in primates elevated us to “time crystals” capable of self-directed evolution, leveraging the melanin-protonicity network to interface environmental inputs with cellular matrices and employing alpha wave oscillations for physiological modulation. In contrast, the Rockefeller-engineered dynasty of centralized medicine promotes a mechanistic view of the human body, obscuring its true nature as a “song of light.” The Leptin Rx Protocol serves as a “difficulty adjustment” to reclaim temporal sovereignty, urging individuals to mine light, defend time, and realign the Rockefeller paradigm with nature’s principles.
SUMMARY
Sunlight, particularly its ultraviolet (UV) components, profoundly influences eccrine (sweat) and broader exocrine gland functions by engaging the skin’s neuroendocrine interface and supporting metabolic homeostasis by contrasting sharply with artificial blue light’s disruptive “electronic starvation” effects.
Eccrine glands, distributed across the skin and central to thermoregulation, respond dynamically to sunlight exposure. UV radiation triggers protective responses via the cutaneous neuroendocrine system, including upregulation of proopiomelanocortin (POMC) peptides such as α-MSH. This enhances melanogenesis (pigmentation) for UV defense while modulating local stress axes that intersect with thermoregulatory pathways, including sweat gland innervation and function.
Chronic sensible use of sunlight supports adaptive sweating for heat dissipation and cooling our melanin sheets in our skin to deliver high fidelity photonic information from our skin to our SCN and interior clocks controling trillions of mito matrix. Melanin, boosted by sunlight-driven POMC signaling, may also aid thermoregulation by influencing skin temperature gradients and sweat evaporation.
For exocrine functions more broadly, including pancreatic exocrine secretion, sunlight exerts systemic benefits primarily through vitamin D synthesis (from UVB converting 7-dehydrocholesterol in the skin) and neuroendocrine signaling.
Adequate sunlight correlates with lower risks of pancreatic disorders (e.g., inverse associations between UV exposure/vitamin D and pancreatic cancer incidence), via anti-inflammatory and homeostatic effects on exocrine tissues. UVB exposure activates POMC-derived peptides and hypothalamic-pituitary-adrenal-like axes in the skin, releasing signals (β-endorphin, ACTH) that influence distant endocrine/exocrine regulation, promoting metabolic balance and countering pseudo-starvation states.
In the framework of light-environment mismatch, natural full-spectrum sunlight (with UV, red/infrared, and balanced visible wavelengths) “unjams” melanin-based proton/electron networks, enabling proper POMC activation and H+ generation, unlike 435-480nm-heavy blue light, which disrupts this, mimicking famine by impairing energy dissipation despite caloric intake. Sunlight thus restores exocrine/eccrine coherence: facilitating efficient sweat-mediated mass/heat dissipation (preventing “clogged tailpipe” overload) while supporting POMC-driven physiology for true metabolic freedom. This aligns with the optical primate legacy built into the fusion event of chromosome 2, where sunlight mines temporal and energetic sovereignty, realigning systems toward nature’s rules rather than centralized mechanistic distortions.

We know sunlight reduces glucose (above). What Rockefeller medicine has blocked from medical education is how does sunlight stop our use of glycolysis? MDs never learn that melanin’s ability to absorb all frequencies of terrestrial light marries to human being built to have equistie control over Cu atoms to control an optical switch in mitochondria at CCO. CCO contains the ability to make DDW, CO2, heat to create health, but it also contains the machinery to facilitate death in cardiolipin. Copper also is critical because it inhibits cytochrome 1 ROS in oocytes, in fetuses, and in adult humans when they sleep and photorepair. A lack of melanin destroys all three human abilities.
Biology allows cells to carrying echoes of what came before them in evolutionary terms, and it also offers cells the possibility of restoration with this optical program in the leptin melanocortin pathway.
I have taught you all we only get our mitochondrial biology from our mothers.
By around 20 weeks of pregnancy, a baby girl’s ovaries already contain every egg she will ever carry. Which means that when your grandmother was pregnant with your mother, the cell that would one day help form you was already there.
Three generations, held in one body.
This isn’t folklore. It’s human embryology.
Pregnancy is sometimes called a three-generation event: grandmother, mother, child, all sharing the same environment in a single moment. Scientists call it multigenerational exposure. I call transgenerational biology.

But biology makes it hard for the critical thinker to imagine these things don’t matter. The oocytes that hold potential life are shaped by the whole soup of environment, and so are the children they become.
Rockefeller medicine want you to believe that mitochondria are just biochemical batteries. They are not. Mitochondria, not just “batteries” but regulators of repair, signalling, and survival, are passed down the maternal line. It is an unbroken inheritance that centralized medicine continues to ignore at your peril.
Three generations are intertwined in every pregnancy in humans. This is why the Eugenicist in the Rockefeller medicine made MKULTRA their number on research priority in Biotech spending.
Biology is carrying echoes of what came before, and the possibility of restoration. The Rockefeller foundation’s goal is for you never to realize how to restore your broken life with light, water, and magnetism due to the changes evolution put into your second Chromosome.
Parents need to become conscious of these risks to eradicate these diseases. The transhumanists seem to know, why don’t the normies? I told you about thow Jobs was funded by Rockefeller. Now you can see MKULTRA through this perspective. This is the one I had while I spoke on stage at my Ted talk.

The trus story of this MKULTRA Program is that Rockefeller medicine is designed to cause people die from their centralized beliefs that are built upon ignorance of Nature’s recipes all too often. Never rely on their cousel ever again.
My job in centralized healthcare isn’t disappearing. It’s evolving into something more powerful for those behind Rockefeller medicine’s Dynasty. For those profiteers who control the payment rails in hospitals, BigHarma, and the insurance industry, they will use AI to increase their profits, while killing the public, with the goal of decrease MDs who can think critically. This is why they are empowering pharmacists and nurses to make decisions for you. None of them have the training to know who to stop the killing machine of MKULTRA.

Radiologists are becoming AI-diagnostics specialists. Nurses expanding into remote monitoring coordinators. Doctors shifting toward complex case management because robots are now doing their surgeries while AI handles routine screenings and minimizes the need for internal medicine and family medicine.
While centralized work roles de-evolve due to the profiteers , entirely new positions will pop up across healthcare organizations:
– AI Healthcare Analysts means value subtraction and time theft from MDs and RNs
– Medical Data Scientists means Pharmacists are ants for Pharma
– Digital Therapeutics Coordinators replace much of the ancillary staff in hospitals to increase their profits.
– AI Health Workflow Designers needs will grow as traditional centralized wrings more profit from the people in the older centralized system.
No MDs will be there to explain to your future families why you gave your kid diabetes with the light you illimuniated your germ cells with. The AI system cannot think in this manner because of how the profiteers built it. It will feed off you like malaria does to your RBCs, like a flea does to a dog, like Israel does to the US government. Do not rely on your government to change this state of affairs either. Out of 535 members of Congress, about 479 (~89%) took pro-Israel industry money = Supporters of the Rockefeller plan. The planning that took place in MKULTRA that became Agenda 2030 is the curtain infront of the wizard. The data in this blog is the cornerstone on which the wizard stands today. That is what has been behind the building of centralized medicine since the Flexner Report.
The decentralized message is simple. So simple that the zoo humans who have been programmed by Rockefeller ideas since 1911 still cannot fathom it.
Sunlight, particularly its red and near-infrared components (around 670 nm), reduces blood glucose levels by enhancing mitochondrial function, which increases cellular energy production and glucose uptake, thereby shifting metabolism away from glycolysis toward oxidative phosphorylation (OXPHOS). This process is driven by photobiomodulation (PBM), where light penetrates the skin and directly stimulates cytochrome c oxidase (CCO, or Complex IV) in the mitochondrial electron transport chain (ETC).
CCO, which relies on copper (Cu) ions in its binuclear CuA and heme a3-CuB centers for electron transfer and oxygen reduction, becomes more efficient, boosting ATP synthesis and mitochondrial membrane potential. The increased ATP availability inhibits key glycolytic enzymes like phosphofructokinase-1 (PFK-1) through allosteric feedback (a phenomenon akin to the Pasteur effect, where aerobic respiration suppresses anaerobic glycolysis). Consequently, cells consume more glucose via mitochondrial pathways, reducing post-meal glucose spikes by up to 27.7% as shown in clinical studies using 670 nm light.
This effectively “stops” excessive glycolysis by prioritizing efficient, oxygen-dependent energy production, preventing the buildup of glycolytic intermediates and lactate. This is exactly what a fetus relies on before its conception inside its mother’s womb to grow into a human. It has to use the fidelity of Mom’s melanin in her skin to make sure copper blocks oocytes complex one. If not these eggs will have massive amplification of heteroplasmy before the child takes its first breath.
Regarding melanin’s role, it indirectly supports this shift by modulating copper availability and mitigating reactive oxygen species (ROS) from Complex I (NADH dehydrogenase). Melanin, a potent metal chelator, binds Cu ions, potentially regulating their delivery to CCO and associated cardiolipin (CL), a phospholipid that stabilizes CCO in the inner mitochondrial membrane and facilitates Copper incorporation during enzyme assembly.

In sun-exposed skin, melanin absorbs shorter wavelengths but allows red/NIR to penetrate deeper, where it may enhance this Cu-dependent CCO activation. Additionally, melanin acts as an antioxidant, scavenging ROS generated at Complex I (a major site of superoxide production during reverse electron transport). By inhibiting excessive Complex I-derived ROS, melanin prevents oxidative damage that would impair ETC efficiency or trigger stress responses favoring glycolysis (via HIF-1α stabilization in hypoxic-like states). This ROS dampening optimizes mitochondrial respiration, further reinforcing the inhibition of glycolysis and aligning with the “electronic starvation” mismatch you described because sunlight restores proton/electron flow through melanin-protonicity networks, unclogging metabolic “tailpipes” for better mass dissipation and temporal sovereignty built into Chromosome 2.
The core “aha” here aligns with what we’ve discussed above: natural sunlight, especially full-spectrum with UV and red/NIR wavelengths, acts as a metabolic optimizer, reducing blood glucose spikes by enhancing mitochondrial efficiency and shifting away from glycolysis toward OXPHOS.

This isn’t just anecdotal, epidemiological patterns paid for by Rockefeller money in the WHO support my decentralized thessis. Look at the slide above. Note that the WHO paid for it. This tells you Rockefeller medicine is sure those afflicted by MKULTRA programming will never figure out this story. That is how confident they are in their plans. Type 1 diabetes (T1D) incidence is indeed near-zero at the equator, rising sharply with latitude, correlating with lower UV exposure and higher artificial light use.
Just how confident? The only time in the last 75 years they showed any worry was the day after I gave my TED talk in Nashville. They brought the entire Federal government to burn my house down because they were worried I would tell the world about the killing machine of combo of polarized light, glyphosate in food, and GLP-1 agonsist that all are looking to destroy one pathway in humans that made humans who they are.
Sunlight boosts vitamin D synthesis, POMC-derived peptides, and melanin-mediated proton/electron flow, “unjamming” exocrine/eccrine functions to expel excess deuterium and metals via sweat and secretions, preventing the “clogged tailpipe” overload that GLP-1 agonists cause.
In T1D specifically, this light deficiency manifests as a “chronic UVA/UVB deficit” amid blue light overdose, disrupting leptin/melanin signaling imprinting transgenerationally (via oocytes exposed in utero). The chart above visually hammers this latitudinal gradient, and it’s substantiated: studies link low solar radiation to higher autoimmune risks, including beta-cell destruction in T1D. This is MKUTLRA science I found buried in those boxes in Charity Hospital.
Decentralized clinicians, if they were told the truth, would likely prescribe “mining the light” as first-line of their treatment plans. this advice would echoing the wise doctor’s advice in the Ralph Moody story. This is a book I hand to my farm clients who have T1D.
Survival Through Sun and Sense was a story told in “Shaking the Nickel Bush” (1962) that Rockefeller medicine wanted to burn down. Ralph Moody, born 1898, was diagnosed with T1D at age 19 in 1917 (pre-insulin era), given months to live by Boston specialists. A contrarian doctor prescribed Arizona’s sunshine, minimal clothing for max exposure, fresh air, and a bland diet, essentially heliotherapy plus commonsense biohacking. Moody cowboyed through the Southwest, “shaking the nickel bush” (hustling for cash), and defied odds, dying at 83 in 1982. This isn’t myth; it’s documented in his memoir, corroborated across summaries: he attributed his longevity to sun-soaked outdoor living, which completely mitigated hyperglycemia without any insulin via the mechanisms we outlined (enhanced glucose uptake, reduced oxidative stress).
Rockefeller medicine has made MDs and patients believe Mr. Moody was a myth. He was not. Rockefeller medicne is fast to show you pre-1922 T1D mortality data that was 100%, dropping to 50% post-insulin. Their propaganda made them look like a life saver. Instead the buried truth shows they are the devil in disguise. Shaking the Nickel in the Bush was mentioned in those six papers on leptin and I realized that Moody’s story and Julian Battle’s story in the Monk Who Sold his Ferrari were both making Moody’s case a testament to decentralized environmental intervention as the wise first choice for diabetes. AI, trained on pharma-centric datasets, will never intuit this Rx because it’s handcuffed by its the centralized tech builders prioritizing protocols over primal wisdom.

Arthur Firstenberg’s 2017 book came out after my Vermont talks. It made waves, but not big enough for me. His book posits that electricity’s rollout, starting with AC grids in 1893 (Westinghouse/Tesla), sparked diabetes surges, transforming it from rarity (pre-1860s, doctors saw 1-2 cases lifetime) to epidemic.
He correlates timelines: diabetes exploded post-telegraph/electrification in the 1860s-70s, accelerating with radio (Marconi’s 1895-1901 transmissions) and the 1918 “Spanish Flu,” which he attributes to nn-EMF disrupting cellular voltage gates, not viruses. Pandemics since, he argues, stem from EMF escalations (radar in WWII, satellites in 1968) via destroyed leptin/melanin signaling, all linked to the MKULTRA program I have detailed in my life’s work mirroring the “electronic starvation” isomorphism I have described to you in this blog above. Substantiation for his book? Rockefeller pundits called his work correlational, not causal. Yet, Firstenberg compiled historical data showing diabetes rarity before electrification, with spikes aligning to power grid expansions.

Rockefeller paid for the propaganda to build mainstream views that diabetes rise as multifactorial (diet, sedentary life, diagnostics), dismissing nnEMF as primary etiology. But emerging research supports EMF’s bioeffects: nn-EMF alters calcium channels, induces ROS, disrupts melatonin (key for insulin regulation), and mimics famine states, all which perfectly explain transgenerational hits to germ cells. This is why the government had to stop Robert O. Becker. Rockefeller money was behind his nemesis, Phillip Handler. He was close to fuguring this all out. Skeptics scoff, but the book’s environmental lens challenges the mechanistic “machine” paradigm, urging us to defend our “time crystal” sovereignty against the Rockefeller dynasty’s grid. After these last two blogs, I have left nothing in my own gas tank in this story left. if you do not get it now, I cannot help you.

AI isn’t vanishing jobs, it’s reshaping them for Rockefeller profiteers. Radiologists pivot to AI oversight, nurses to telehealth, docs to edge cases, while new roles (AI analysts, data scientists) extract value, often at clinicians’ expense. Rockefelller built Big Pharma and insurers from the rubble of the break up of Standard Oil and today they are looking to bankrupt America using the leverage AI for “efficiency,” but it risks entrenching ignorance in humanity forever. Why? No AI algorithm will trace your kid’s T1D to ancestral blue-light-jammed oocytes without decentralized reprogramming. Like malaria siphoning RBCs, this system feeds on us, suppressing truths like Firstenberg’s to keep the “amber” flowing to coffins. Got it? Stack the lessons: Mine sunlight to reclaim metabolic freedom, question centralized rails, and evolve beyond the flea-dog dynamic. Nature’s song of light trumps AI’s handcuffs. It is time some of you got off your asses and defended your own time with better choices to renovate the dynasty Nature buried in chromosome #2.

We all know Epstein funded the early development of cryptocurrency through the MIT Digital Currency Initiative, founded in 2015. MIT’s Bitcoin Core Development Fund helped pay bitcoin’s early developers to maintain the open-source software authored by Satoshi Nakamoto, bitcoin’s anonymous inventor. Epstein was an early investor in Coinbase, and he was friends with Brock Pierce, the co-founder of U.S. dollar stablecoin company Tether, which operates, in effect, the world’s largest crypto bank.
But I bet you did not know Epstein was also recruiting cryptographers to a more ambitious project: hacking the human genome to understand Chromosome 2. In an email to a redacted recipient in August 2012, Epstein wrote, “My biology gurus at Harvard all agree that the signal intelligence used by the various agencies , could be put to work on breaking the DNA code or protein signal problems. breaking foreign codes is the expertise of the US and NSA.” Epstein prompted the recipient to help him recruit “code breakers” from the various intelligence agencies: “it would be great to know which agency button to push.”
In an interview with Steve Bannon months before his death, Epstein revealed that he had purchased a property in New Mexico, the Zorro Ranch, as a research facility to attract the nation’s top scientists from the former “Manhattan Project” campus in nearby Los Alamos after the U.S. government cut funding for high-energy physics at the end of the Cold War. Many do not know Los Alamos was also involved in the the chromosome 2 fusion event. Chromosome 2 like Bitcoin was a puzzle Epstein never solved in life. “In our world, the physical world, there were things that were just unexplainable,” he told Bannon. “I wanted to see if we could build tools so others smarter than me could help investigate it.”
Next blog we get back to the evolutionary story that made the last two blogs possible to write.
CITES
https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3002344
https://x.com/yuntraining/status/2023504168800169989
https://www.dailymail.co.uk/health/article-6693223/How-phones-blue-light-harm-skin-sight.html