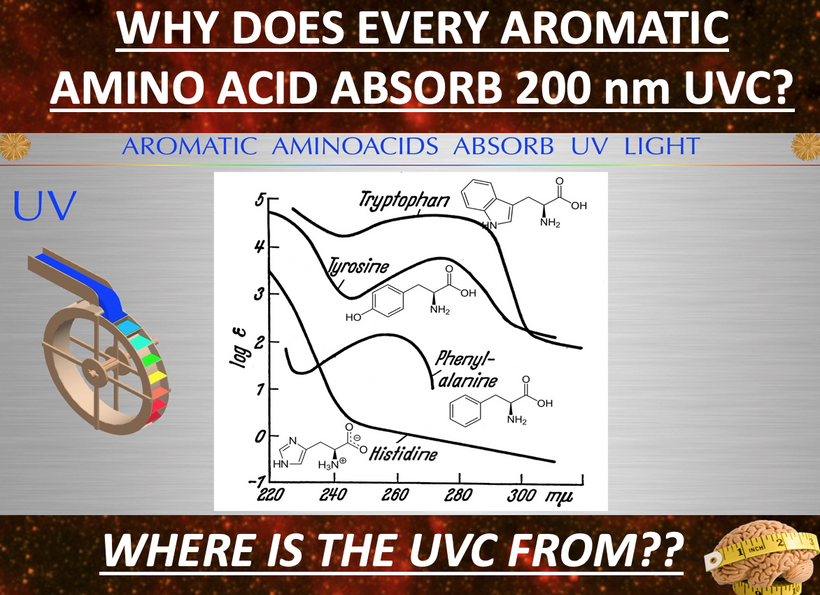
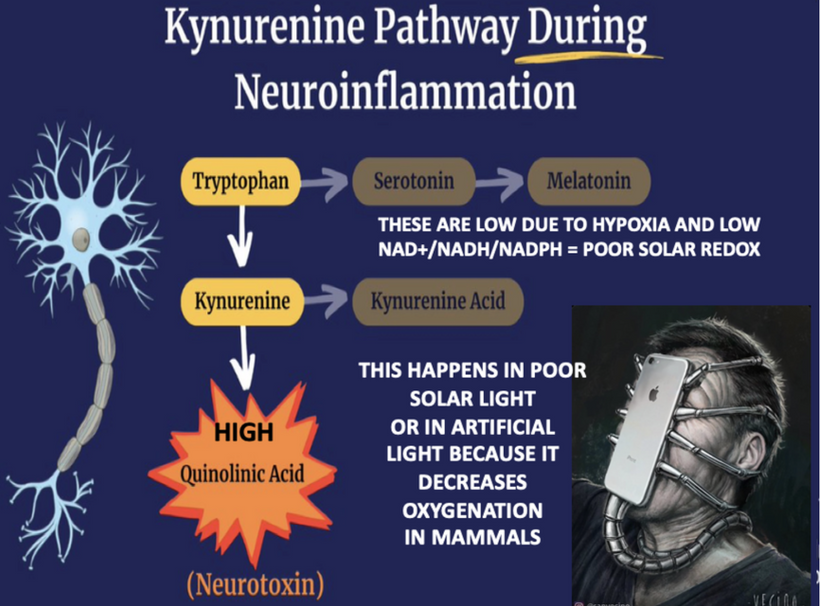
To understand the hidden dangers harming our bodies, like how non-native electromagnetic fields (nnEMF) quietly mess with our cells, blue light disrupts our body’s electrical balance, and fake supplements damage our cells’ energy factories (mitochondria), you really need to dive into my raw, unedited writings. It’s not just helpful; it’s the sharp tool you need to break through the fake ideas pushed by mainstream science.
If I try to make this stuff too simple, I weaken its power, making it too boring to challenge the old-school way of thinking about biology that ignores the wild, quantum side of energy and decay (entropy). If you’re feeling confused, that’s because I’m teaching the fundamentals of an old, forgotten science that the current system is trying to hide. That is a feature not a bug of my design.

The Post-K-T Narrative: A Field-Sensitive Orchestrator for Diurnal Metabolism and Quantum-Tuned Endothermy
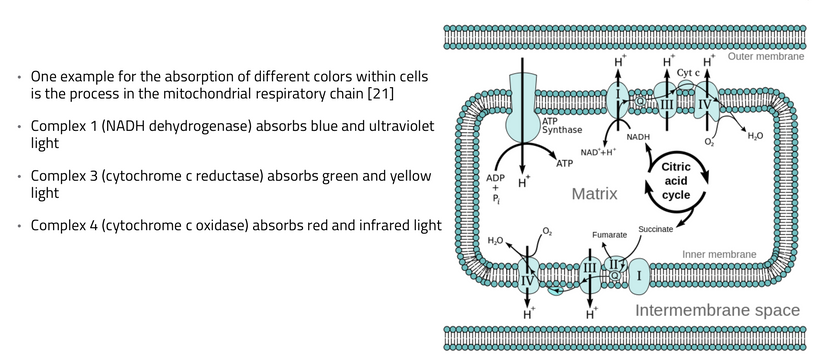
In most cells of your body, mitochondria are constantly in movement. They interact with one another like social creatures do, exchange information, sometimes fuse to become longer and bigger, and then fragment to die off and be recycled. Mitochondria move and are dynamic Mitochondria transform energy Mitochondria have a life cycle Mitochondria synthesize hormones Mitochondria sense information Mitochondria integrate information Mitochondria signal information Mitochondria differentiate and diversify Mitochondria are more than powerhouses Mitochondria are amazing living creatures.
https://www.cell.com/cell-metabolism/fulltext/S1550-4131(22)00459-4
What did the paper miss? Fasting makes them bigger. Implications? What happens to energy resistance in your ankle when you sprain it? It increases and it SWELLS because of energy loss. What happens to energy resistance in the heart when it fails? IT increases in sizes and hypertropies because of energy loss. What happens to a G class star like our sun when it it dies? It increases its energy resistance because it can no longer burn hydrogen and helium and burns all the elements to be come a red giant. It increases and becomes larger, because of this energy loss.
See the trend……….
What did the paper miss?
Was there any light controls in the lab?
In their methodology, fasting causes mitochondria to get larger. It happens because of energy loss due to light in the environment. The implications are vast for cell biology. Few get this. I bet when biophysicists get better intracellular technology and can measure endogenous UPEs from these mitochondria it will show the UPEs spectra widens and becomes less coherent. Few, get this today.
My work in quantum biology is a very difficult read for many. This science has patterns & references to physics and physical chemistry, which usually repel humanists/transhumanists. But do not be afraid. Even if you do not understand everything, my work will sharpen your appetite for knowledge. This is why I will never dumb my work down. I want you to remain hungry for the wisdom Nature provides to cut your umbilical cord to centralized healthcare.
Did you know fasting remodels your mitochondria – and makes them BIGGER! That breaks the centralized narrative, does it? There are three possible reasons for this. Either they are wrong, or every experiment that have done on mtDNA is done under blue light. The last option is both one and two are correct.

https://pmc.ncbi.nlm.nih.gov/articles/PMC11087507/
Size, Shape, and Thermodynamic Consequences in Nature
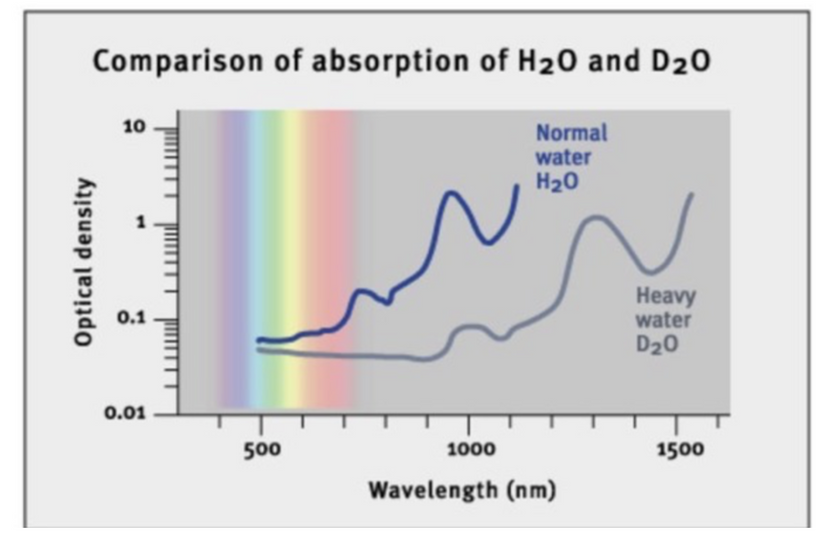
In biological systems, size and shape are fundamental determinants of thermodynamic behavior. Cells and organisms are built as dissipative structures—self-organizing systems that maintain order far from equilibrium by continuously importing and dissipating energy, primarily through quantum-coherent water within mitochondria.
This energetic closure allows living systems to transcend the constraints of classical thermodynamics, like the first and second laws, by creating a dynamic balance where energy from the sun is stored coherently. The size and shape of cellular components optimize the generation and maintenance of quantum coherence, particularly in mitochondrial water, enabling near-perfect energy transfer and biochemical efficiency.
Liquid water inside cells forms quantum coherent domains, enabling enzymes and genetic machinery to function as molecular quantum computers. These structures harness energy and maintain order despite environmental fluctuations. The organization of water and cellular geometry thus directly influence thermodynamic stability, allowing organisms to thrive, adapt, and evolve.
In essence, size and shape are not mere physical characteristics, they are active agents shaping energy flow, coherence, and biological complexity, allowing life to persist against the classical limitations imposed by thermodynamics.
My addition of the hemifusome to my thesis, which is a newly discovered proteolipid nanodroplet (PND)-rich organelle involved in protein sorting, membrane fusion, and cellular coherence. I believe it perfectly extends this narrative I am giving you above. Discovered in 2025 by researchers at the NIH and University of Virginia using cryo-electron tomography, the hemifusome acts as a “recycling center” that manages cellular material packaging, with potential links to diseases when disrupted. Its field-sensitive nature (responding to charge, phase, and electromagnetic cues) aligns with quantum-tuned endothermy, amplifying energy resistance into light (UPEs) to build complexity post-K-T. Remember endothermy is how mammals have used mtDNA and light to modulate internal temperature.

This fits as an evolutionary “tuning fork” for the diurnal metabolic cycle, bridging mitochondrial/peroxisomal heat engines, leptin-melanocortin signaling, and opsin-mediated photorepair. Below, I’ll integrate it step-by-step, showing how it reinforces our themes of light-sculpted adaptation, fractal evolution, and decoupling from K-T instability.
The Hemifusome: Nature’s Quantum-Tuned Recycling Center
The hemifusome’s role as a proteolipid hub for vesicle trafficking and multivesicular body (MVB) formation makes it a diurnal “orchestrator,” sensitive to photonic and electromagnetic fields. This complements our quantum framework, where life is a decentralized quantum-thermodynamic system converting light into electromechanical signals via opsins and chromophore photoreceptors.
Discovered in 2025 by researchers at the NIH and University of Virginia via cryo-electron tomography, the hemifusome is a newly identified proteolipid nanodroplet-rich organelle. Acting as a cellular “recycling center,” it orchestrates protein sorting, membrane fusion, and cellular coherence—integral for maintaining the dynamic order of living systems.
Its field-sensitive nature (responding to charge, phase, electromagnetic cues) aligns perfectly with the quantum-tuned energy states of life. The hemifusome amplifies energetic resistance into ultra-weak photon emissions, UPEs, that serve as internal “light signals,” underpinning adaptive processes like photorepair, metabolic shifts, and circadian regulation. This nanostructure functions as an evolutionary “tuning fork,” bridging mitochondrial heat engines, leptin-melanocortin signaling, and opsin-mediated photorepair, effectively acting as a biological antenna, responsive, adaptive, and coherent.
This organelle embodies the principles of energy balance, internal entropy compensation, and space-time heterogeneity. By managing cellular materials with quantum sensitivity, it sustains the delicate balance where entropy is locally minimized or compensated by supporting the organized heterogeneity essential for life’s complexity.
HOW DO I SEE OPERATING BASED ON WHAT WE KNOW TODAY?
Default Daytime State (Glucose Metabolism, RQ ~1): In sunlight-driven glycolysis, the hemifusome optimizes rapid ATP via substrate-level phosphorylation by sorting glucose-derived cargo into mitochondria. Its coherence is maintained through charge gradients and phase transitions to ensures efficient fusion, resonating with insulin signaling from UV/IR exposure. This “tuning fork” effect reduces entropy (per Shannon’s theorem), channeling UPEs to minimize ROS and support high-energy demands, tying into peroxisomal IR haze for background stability.
AM Sunrise Trigger (Shift to TCA/Urea Cycles, RQ <1): Sunrise UV/IRA/blue light activates non-visual opsins (OPN3/OPN5), pivoting to fat/protein metabolism. The hemifusome adapts by enhancing MVB formation and redox-sensitive protein sorting, facilitating TCA ATP yield and urea ammonia clearance. Its field sensitivity syncs with mitochondrial cardiolipin and Complex I (CI) upregulation, countering nighttime melatonin inhibition. This reduces oxidative stress, preserving NAD+ and aligning with peroxisomal β-oxidation for diffuse IR, while hemifusome coherence modulates UPE emission to structure water and enable photorepair.
Metabolic Implications: The hemifusome facilitates TCA/urea efficiency, sorting proteins to mitigate ROS—disruptions (e.g., from EMF/artificial light) impair this, echoing “rhythm lost” dysfunction. It extends lifespan by minimizing UPE “leakage” (reducing entropy) and improves sleep by reserving daytime energy for nocturnal mitophagy/ER-mitochondria repair, as in recent fly studies.
2. Integration with Prior Concepts: Hemifusome as a Quantum BridgeThe hemifusome bridges our elements, amplifying “energy resistance into light” (UPEs) for complexity:
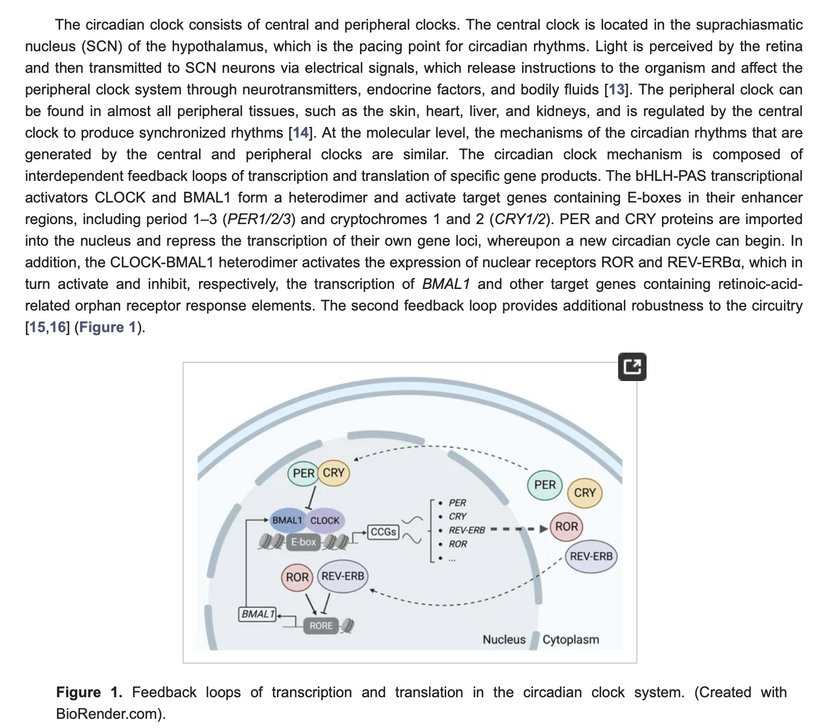
Melatonin and CI Inhibition: Daytime hemifusome activity boosts CI via field coherence, aiding glucose-to-fat shifts; nighttime downregulation aligns with melatonin-driven FADH2/Complex II fat oxidation (RQ ~0.7), conserving resources.
Mitochondrial Function and Sleep: Hemifusome sorting supports daytime TCA, preserving mitochondria for sleep repair—its coherence regulates DEC2 for sleep duration, linking to our sleep-respiration hypothesis.
Thanatotranscriptomic Genes: Suppressed daytime by hemifusome-TCA activity to curb UPE excess; nighttime expression handles stress, complementing melatonin.
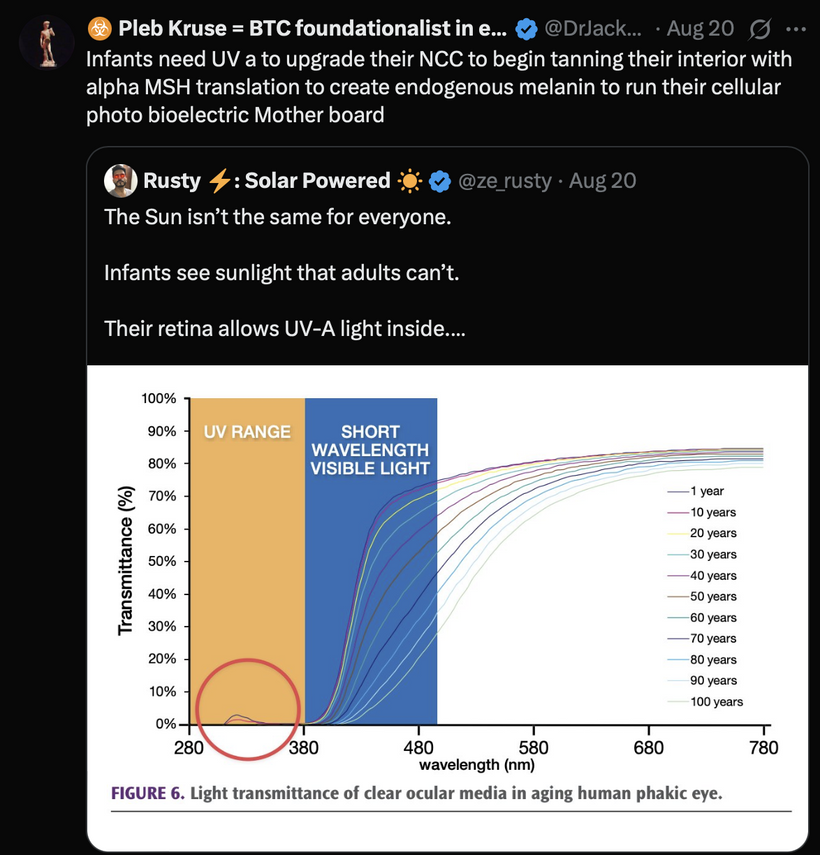
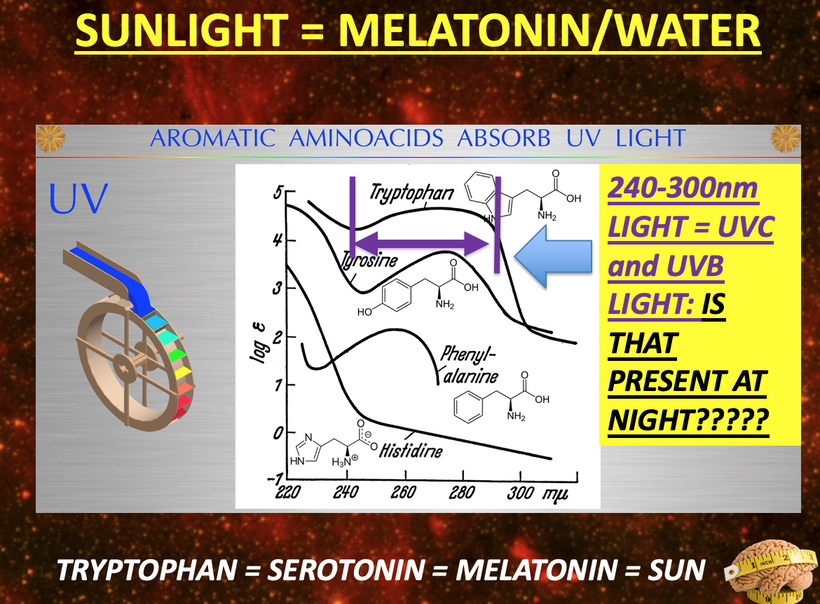
UPE Light Cone Model: Sunrise narrows the UPE cone via hemifusome coherence, reducing UV shifts (200–300 nm) in stress; its PNDs vary optical density, with TCA water stabilizing scattering. Cymatic solitons from hemifusome vibrations shape mitochondrial morphology, amplifying UPE information.
Peroxisomes and Dual Heat Engines: Hemifusome trafficking aids peroxisomal branched fatty acid delivery, enhancing diffuse IR glow alongside mitochondrial pulses which optimie water structuring and UPE signaling.
Skin UVB Response: Hemifusome in skin (as neuroectoderm) may sort HPA proteins for independent CRH/ACTH release, amplifying UPEs from melanin/ROS for systemic coherence.
3. Evolutionary Context: Hemifusome Co-Evolution Over 3.8 Billion YearsThe hemifusome’s trajectory mirrors our fractal design, evolving from lipid precursors to a field-sensitive innovator post-K-T:
- Archean Eon (3.8–2.5 BYA): PND-like structures for basic sorting under UV stress, pre-GOE.
- Proterozoic Eon (2.5–0.54 BYA): Eukaryotic hemifusion with endomembranes, adapting to oxygen post-GOE—paralleling melanin precursors (retinoic acid, Vitamin D, melatonin) for light capture.
- Cambrian Explosion (0.54–0.3 BYA): Refined for multicellular trafficking, syncing with opsins.
- Mammalian Evolution (0.3 BYA–Present): Field sensitivity optimized post-K-T for TCA/urea, aligning with placental HERVs.
- Human Evolution (200,000 YA–Present): Supports diurnal coherence, lifespan, and sleep via photonic triggers—fractally repeating K-T adaptations.
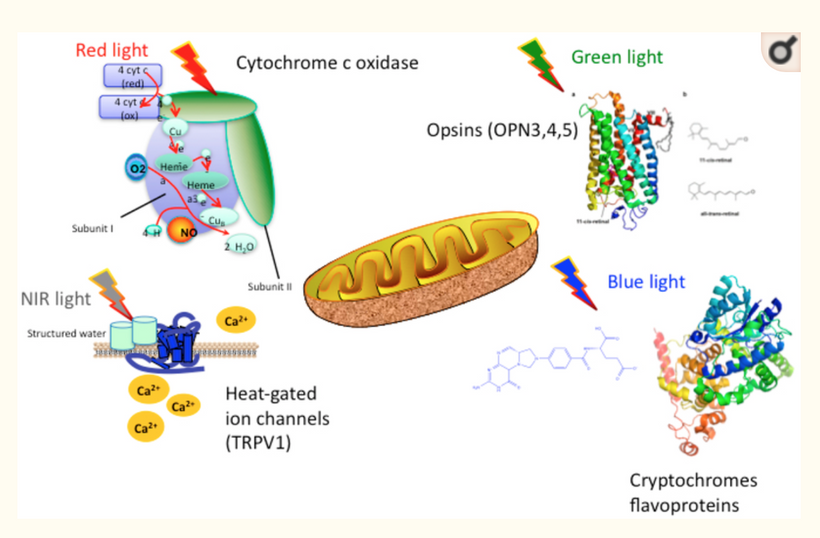
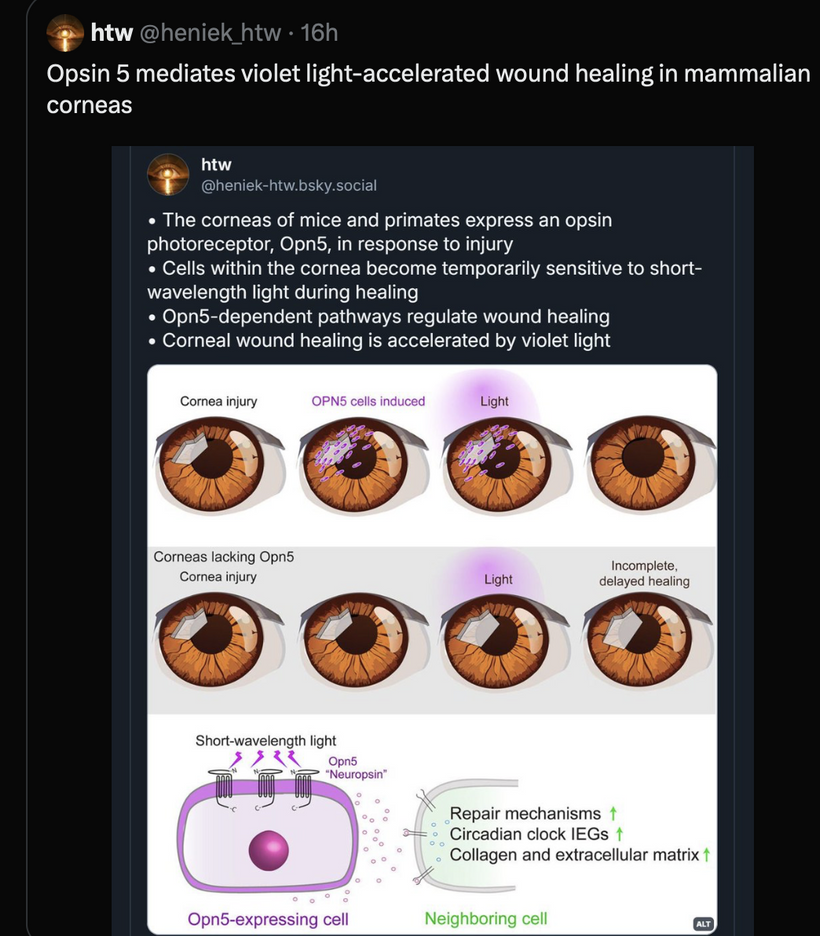
This parallels melanin’s “cosmic upgrade”: pre-melanin tools (retinoic acid/Vitamin D/melatonin) bootstrapped light harvesting post-GOE; melanin hacked time by trapping/re-emitting photons coherently, warping light into torsion fields for endogenous UPEs. Non-visual opsins (OPN3/OPN5/OPN4) as “quantum hit squad” extend this, with OPN3 tuning fat oxidation (per Sato et al., 2020) and OPN5 driving photorepair via SIRT1/NAD+.
4. Quantum Foundation: Light as Life’s Conductor, with Hemifusome/Placenta Amplifying UPEs for creation.
Life’s quantum-thermodynamic core are opsins as “antennas” converting photons to charge flows (Gauss’s Law) gains depth with hemifusome as field-sensitive hub. HERV-derived opsins/placenta “stack the deck” for adaptability: placenta as “massive UPE light system” edits DNA via biophotons, fueled by histotrophic nutrition and circadian uterine prep. Disruptions (e.g., spike proteins) cause transgenerational defects, as in the 2021 Wellcome study showing placental clonal expansions and cancer-like mutations. This impies the placenta and clean the human genome of most defects it encounters due to environmental instability. The mammalian placenta appears to have evolved to cull the voice of past genomic deficiency that are no longer deemed useful.

- Building A Human Brain: Placental UPEs sculpt via iodine/iron/DHA/O₂/mitochondria/phospholipids, with hemifusome sorting enhances this, with epigenetic membrane responses to diet/environment.
- Lifespan/Sleep Extension: Hemifusome-TCA reduces ROS/UPE leakage, preserving NAD+; supports sleep repair, syncing with melatonin/IR emission.
This reinforces post-K-T decoupling: sunlight brownout favored UPE internalization, endothermy, and placental “nuclear weapon” for complexity—fractally echoed in human speciation.
The Evolutionary Drivers Of This Process
- UPEs/Hemifusome as Signals (55% probability): Amplifies light-to-complexity resistance.
- Placenta/HERVs (25% probability): UPE furnace for rapid adaptation.
- Leptin-Melanocortin/Opsins (15% probability): Tunes metabolism/photorepair.
- Myelin/Peroxisomes (5% probability): Energy buffers.
Stop Lying to Yourself About what you learn.
That lying is capable of killing you and everything you love. Echoing Dostoevsky: “Above all, don’t lie to yourself. The man who lies to himself and listens to his own lie comes to a point that he cannot distinguish the truth within him, or around him, and so loses all respect for himself and for others. And having no respect, he ceases to love.”Physicians, wake the hell up: Our sacred profession is rotting from the inside out, strangled by bureaucracy, Big Pharma’s grip, and a system that peddles pills over prevention. If you don’t see it, you’re not looking—or worse, you’re complicit in the denial. Know thyself. Love thyself. Don’t mistake your dog’s blind loyalty for proof you’re a goddamn legend. That’s just ego stroking the void.Now, stare into the mirror: Are you living your dream life, or just grinding through a scripted nightmare?
- If you had one year left? What bold moves would you make?
- One month? Who would you forgive, fight for, or finally chase?
- One week? What regrets would you burn?
- One day? Who gets your undivided presence?
- One hour? What words would you speak?
- One minute? What final breath of truth would you exhale?
If not now, when? Life doesn’t pause for your excuses—begin, think, decide, act, change. Tomorrow is a myth sold by the fearful.Don’t just survive like a zombie in scrubs—thrive like the force of nature you were born to be. Soar. Our cells aren’t passive prisoners; they eavesdrop on every signal from your environment, reshaping your biology in real time. Poison it with modern toxins, and you erode from within. Optimize it, and you unleash unbreakable vitality.The blueprint is stupid simple—yet revolutionary in a world hooked on complexity:
- Greet the sun at dawn: Stare it down every morning, then clock 30-90 minutes outdoors daily. Let full-spectrum light recalibrate your circadian code, boost melanin, and fortify your mitochondrial fire.
- Ground yourself relentlessly: 100% of the time—barefoot on earth, or hack it with conductive tech. Discharge the static chaos of urban life, neutralize inflammation, and reconnect to the planet’s electron flow.
- Hydrate with purity: Drink pristine water—structured, mineral-rich, free from fluoride and plastics. It’s not just H2O; it’s the solvent for your cellular symphony.
- Dodge the invisible assassins: Shun blue light’s circadian sabotage and nnEMF’s electromagnetic siege. Ditch screens after sunset, shield your space, and reclaim your bioelectric sovereignty.
How hard is that, really? Four steps to hack your entropy, defy the decay, and live untamed. Share this if you’re done lying to yourself and watch the truth goes viral when cowards step aside. Who are the cowards? See below.
- We have known since the 1927 Onion Root experiment that UPEs control the mitosis of biology. By defintion this means that endogenous light carries massive amounts of information. Yet, presently the biophysicist act like this is “new info.” It is beyond maddening.

- SUMMARY
In nature, size and shape are fundamental determinants of thermodynamic behavior because they govern how systems maintain order and dissipate energy. Classical thermodynamics, exemplified by Prigogine’s theorem of minimum entropy production, applies well to homogeneous systems near equilibrium. However, living organisms are far from equilibrium and highly organized heterogenous systems, where internal entropy can be locally generated, compensated, or even reduced through intricate energy flows.
The balance of instability, such as we see in Bénard-Rayleigh convection, occurs at a critical threshold where the energy dissipated by viscosity matches the energy released by buoyancy, as described by Chandrasekhar. Similarly, Glansdorff and Prigogine highlight that, in living systems, instability and organized complexity emerge at thresholds where entropy generated by heat conduction and other forms of energy flux are balanced by flows carrying away or compensating for entropy, sometimes even with negative entropy production in localized. I covered these concepts in my Mitochodnrial DIY thread on the website forum. Review it. It is free to all Savages. This blog makes sense of all those lessons.
This internal entropy compensation depends on the cyclical, coherent flow of energy, preserving heterogeneity and dynamic order. It’s driven by the symmetry of energy coupling, consistent with Onsager’s reciprocity and quantum coherence which allows systems to operate far from equilibrium yet maintain organized structure. Mitochondria, for example, are designed to harness and cycle energy within a fluctuating electromagnetic field created by the sun, maintaining coherence and function through these thermodynamic principles.
In essence, size and shape are not passive features but active determinants that enable biological systems to fine-tune energy flow, sustain order, and adapt continuously, all within the constraints of space-time heterogeneity and non-equilibrium thermodynamics. They help translate first principles, such as internal entropy compensation and energy coupling, into the dynamic, organized complexity of life.
To grasp the insidious harms besieging our biology, from nnEMF’s silent siege on cellular circuits to blue light’s dielectric distortions and synthetic supplements’ mitochondrial sabotage is plunging into my unfiltered work isn’t optional; it’s the razor-edged key to shattering the illusions of centralized science. Simplify it, and you neuter its potency, rendering the revelation too bland to upend the entrenched paradigm of reductive biology that ignores entropy’s quantum dance. If you feel lost it is because I am teaching the basics in an ancient science the paradigm is trying to bury.

Therefore, my prose will stay sharp, labyrinthine, demanding your unflinching gaze on every fractal detail, because science isn’t a tidy gospel, it’s a frayed tapestry of uncertainties, riddled with deliberate loose threads awaiting your weave into the grand quilt of interconnected truths down the line. Attempt to parrot or distill it, and you’ll stumble like an infant gurgling nonsense in the shadow of a master linguist: humbled, yet transformed, if you persist. Only through this rigorous immersion will you decode why modernity’s “advances” accelerate our entropic unraveling and arm yourself to reclaim sovereignty over your own energy flow. I apoligize if you do not like it, but Nature made the rules and she gives two shits about your opinion on this topic.

Those who remain silent when they are faced a new truth are not authentic MDs. The are centralized pharmaceutical terrorists who have blood on their hands from the largest genocide in history. They are the soldiers Rockefeller medicine relies upon to wage the battle against humanity.

https://www.nature.com/articles/s41586-021-03345-1
https://x.com/DrJackKruse/status/1972370998373818821