
The title is provocative because contrary to popular belief a high fat diet can make you quite obese when a couple of variable are present that our modern environment favors today.
This blog fully explains why I knew I had to quit being on call and why I had to regain my time at night and weekends to remain well as I age and am afflicted by mitochondrial damage built into us by nature. If I did not test my own biases I would have be facing different challenges then I do today.
THE BLOGS MAIN POINT UP FRONT:
Focusing in on food is losing proposition for the aging human. This sound nuts when you first hear it but is very wise once it is fully explained so you can understand the perspective.
Black Swan mitochondriacs have learned this lesson the hard way. They focus in on the engine and not the fuel to the engine. The analogy is simple. If you want your Ferrari to go 225mph constantly the main thermodynamic variable that will help you attain this goal is making sure the engine works perfectly. If the engine is perfect it can consume 87, 90, or 93 octanes to get this goal at any one time. If you use cheap gas over time it can affect the performance of the engine, IF YOU NEGLECT to maintain the engine maintenance. This brings up an interesting question to think about before I go full nuclear on the science.
Why do so many gurus recommend a high-fat diet to people who are aging, people with mitochondrial damage, or people with mitochondrial diseases?
Is ketosis supposed to be from the foods we eat of the liberation of fat from our own fat mass?
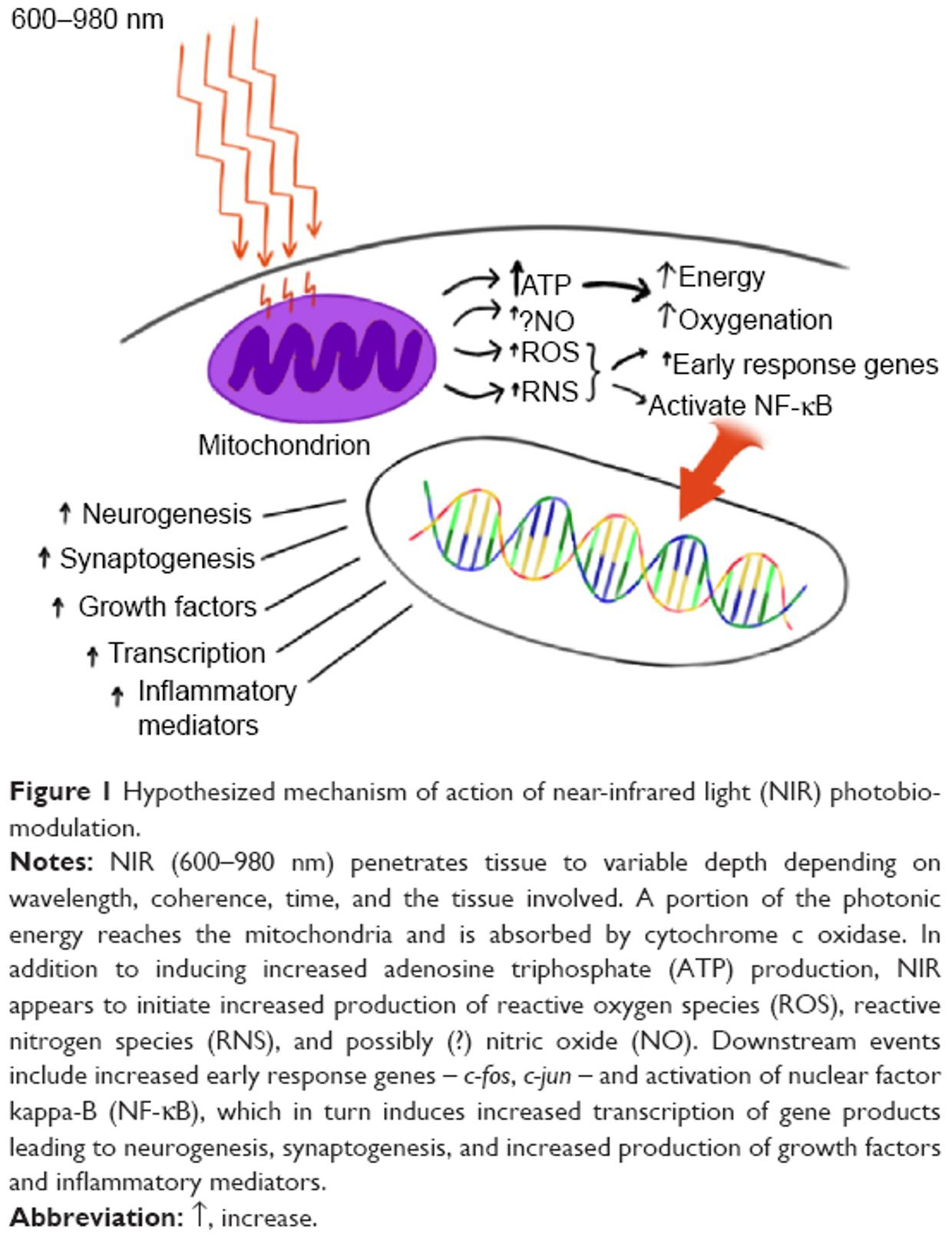
What good is a ketogenic diet from fat if you cannot use it because your matrix is defective? The only way to use fat is via beta-oxidation via the TCA cycle. Oxidative phosphorylation is made possible by the close association of the electron carriers with protein molecules along the inner mitochondrial membrane. The proteins on this membrane are unique, devoid of DHA (a special PUFA), and seem to guide the electrons along the respiratory chain so that the electrons move sequentially from one enzyme complex to another—with no short circuits in normal functioning.

This leads to a stable ROS signal in mitochondria that build complexity using information quanta in electron and proton quantum spin. The ROS signal is a function of oxygen content and the electrons within the inner mitochondrial membrane NOT experiencing any electrical short circuits. Does this situation persist in life? No it does not, it varies as the distance between the proteins change and this is what aging really is. We call this heteroplasmy in biology. Short circuits become more likely and this changes the free radical signals possible while lowering oxygen levels which indirectly forces NAD+ at cytochrome 1 lower.
NAD+ stands for nicotinamide adenine dinucleotide. It has two states, one with electrons and one without. The electrons come from carbohydrates that grow in powerful light latitudes.

Moore circa 2010 sans “massive” technology diet

Moore (2017) on nutritional ketosis running his empire on social media
It has been widely assumed that a ketogenic diet using ketone bodies as a substrate can raise endogenous NAD+ to improve redox status but all these studies were done on nocturnal mammals who have scotopic retinas and skin. This is a very bad assumption considering that NAD+ is a FLUOROPHORE protein with a 340 nm spectral pattern. UVA light makes nitric oxide in our skin, blood vessels in our tissues to affect mitochondrial function.

This is why nature built it to handle excited electrons from foods that grow when UV light is present to a high degree. This error has spilled over to the LCHF and ketogenic groups in humans. For nutritional ketosis to work it has to be coupled with UVA exposure of the eye and skin because it lowers ECT and energy production in the mitochondrial matrix.
If it is done under the power of blue light, the ROS from melanopsin signaling destroys the quantization of NAD+ and NADH at cytochrome 1 to make is highly dysfunctional (picture above). This means mitochondrial damage changes the kinetics of how NAD+ can operate in HUMANS irresepctive of our dietary fuel choices. This means dietary fats can make us quite fat if cytochrome is damaged and IR-A and UVA light are absent from our life.
The recent research cited below in cite 1 (pic above) shows these assumptions might be dead wrong in humans because it appears that the ratio of NAD+ and NADH can vary independently because they are compartmentalized. It turns out NAD+ and NADH are affected by the PHYSICAL location of certain metabolic pathways. Different locations introduce timing and relativity to the equation, and no one seems to realize how this is affected by altered by the external lighting the animal is under to change the kinetic flux of the pathways mentioned in this blog.
For example, the TCA cycle is in the matrix, while the urea cycle is in the cytosol. The place where they meet is called Kreb’s bicycle in research circles. Higher quality research is needed to further identify where and when ketogenic therapy increases the NAD+/NADH ratio in humans. The study needs to be done under both fake light and sunlight to see the real effect on human obesity because of the absorption spectrum in the inflection point of the UVA and UVB part of the visible spectrum of light.
THE INCIDENT EMF CHANGE THE FREE RADICALS MADE IN THE MITOCHONDRIA.
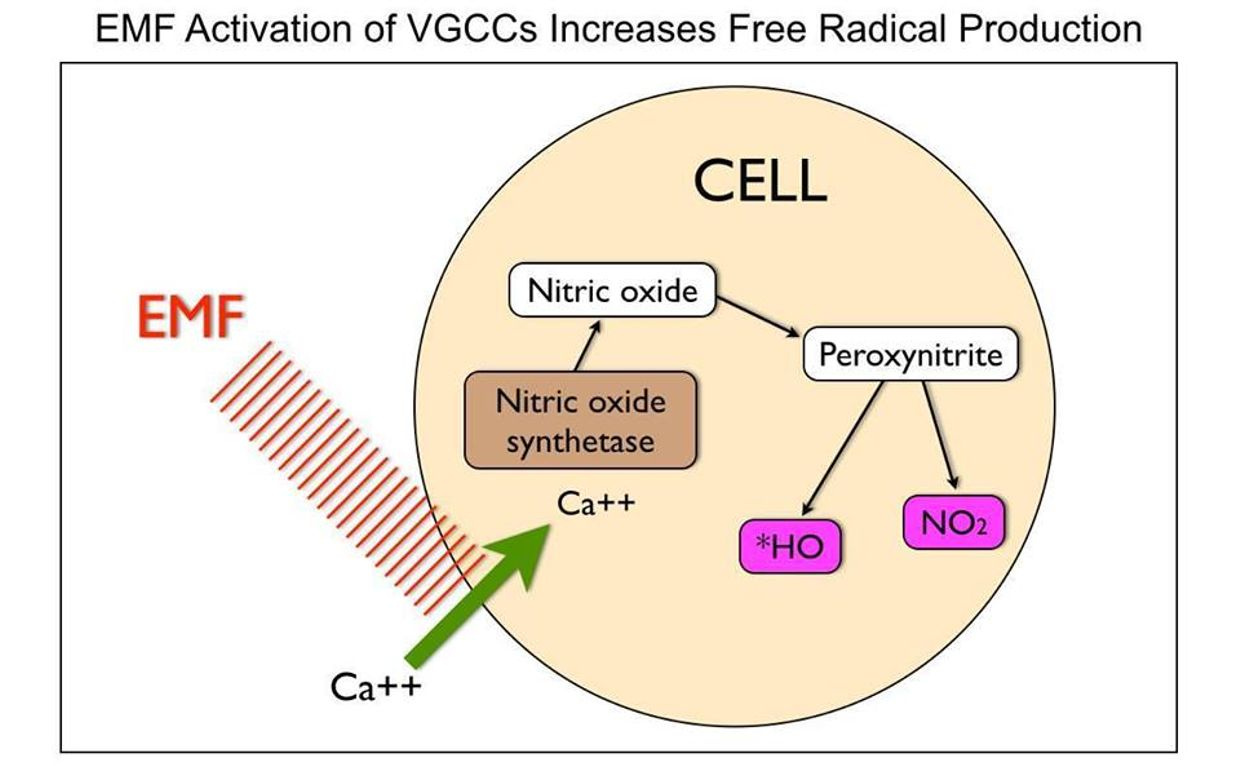
UV-A light makes Nitric oxide (NO). Nitric oxide inhibits electrons motions from cytochrome 3 to 4. It is a breaking mechanism to keep us thin and if you do not get any UVA and replace it with blue light from technology you just gave your body the signal to FATTEN.
These ideas will be critical in delineating specific downstream effects. This implies that a ketogenic diet could be highly fattening to humans who are blue light toxic. Blue light toxicity leads to ROS in the matrix.

There are two type of blue light Hazards known in humans. Noell et al. has described a rhodopsin mediated class 1 blue light hazard (BLH) and Ham et al. categorized a class 2 blue light hazard mediated by lipofuscin. Each cause different collateral effects because of the metabolic pathways they destroy.
Right now I believe the observation of modern humans supports this position fully and this 2018 paper from UT tells me the things I wrote in the Ubiquitination 4 and 5 blogs has even more wind in those sails than I realized when I wrote them years ago. The last cite below shows you how long I have believed that something else beside insulin was behind fattening of humans in technology 24/7.

It will be critically important in future studies to optimize the methodology carefully to ascertain if changes in the NAD+/NADH ratio are caused by changes in NAD+ or NADH solely (unlikely), or as levels of these two redox molecules can also vary independently. This is more likely because of the circadian mechanism at play at cytochrome 1. Supplement makers are now heavily pushing NAD+ supplements because they are betting that raising NAD+ alone varies by its concentration. That is unlikely because its effect is now known to be compartmentalized and is subject to pathway kinetic effects and flux.

Many low carb researchers/gurus have proposed that the decreased reduction rate of NAD+ to NADH during ketone-based metabolism increases the availability of NAD+ and thus alters the NAD+/NADH ratio. There are several problems with this assumption. The first one is that NAD+ is reduced by the addition of hydrogen to make NADH. This happens in the matrix which is a tightly regulated organelle. The hydrogen in this location must come from the hydrogen pool in the matrix. This is not true in all tissues in humans as the picture below shows.
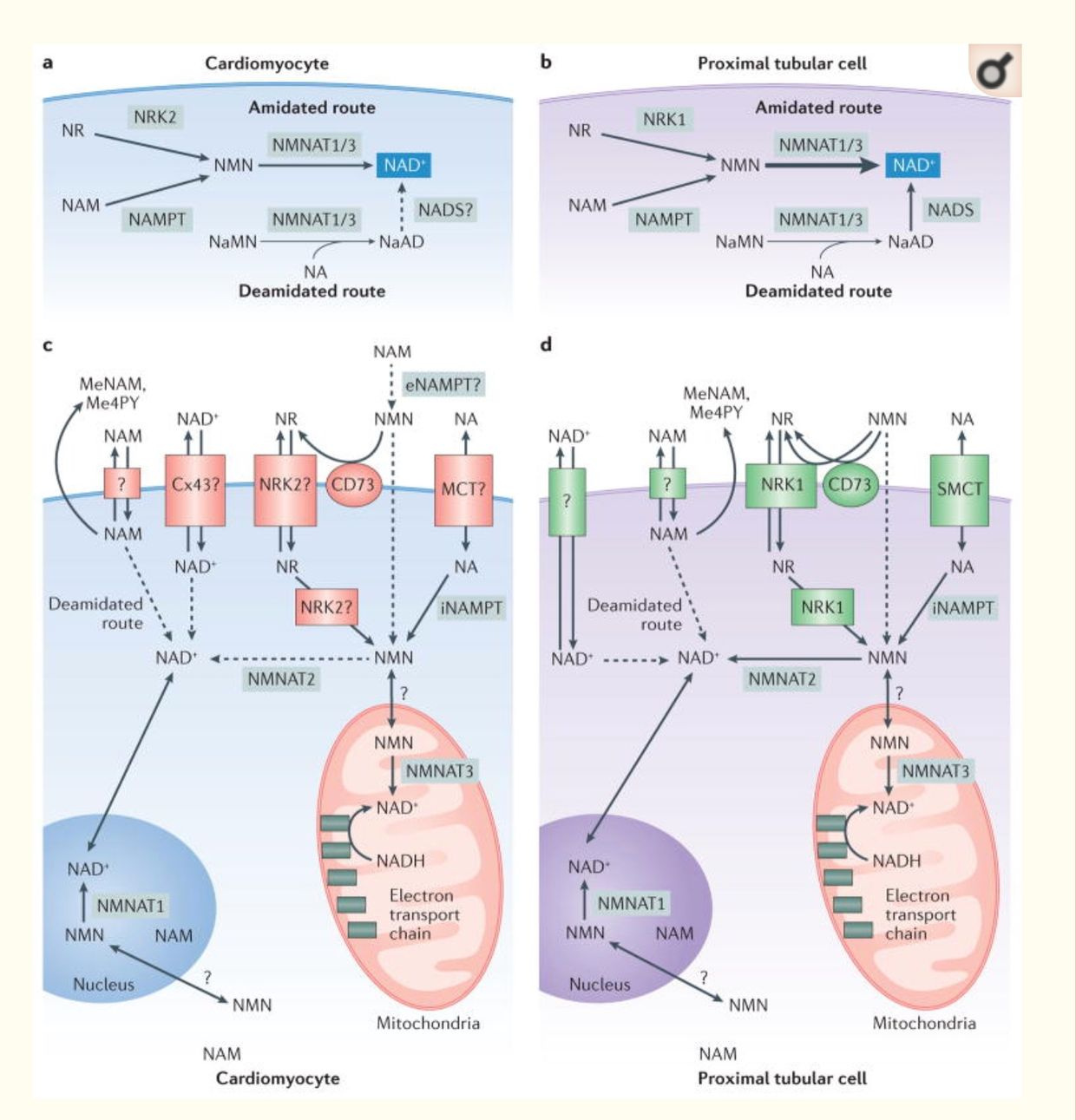
For example, the heart and kidney can use alternative pathways to create NAD+. For example, the de novo synthesis (the deamidated pathway) uses the amino acid tryptophan, which is metabolized to form biosynthetic intermediates. These intermediates ultimately generate nicotinamide (the pyridine moiety of NAD+) and then form NAD+. Another pathway to NAD+ is the use of dietary vitamin B3 compounds, including nicotinic acid, nicotinamide and nicotinamide riboside, serve as NAD+ biosynthetic precursors and are salvaged from the diet (the amidated pathway) to generate cellular NAD+.
Tryptophan works with UV light from 260nm-290nm as the picture below shows. This is big time short wave UV light close to the UVC range. Why doesn’t anyone see these connections?
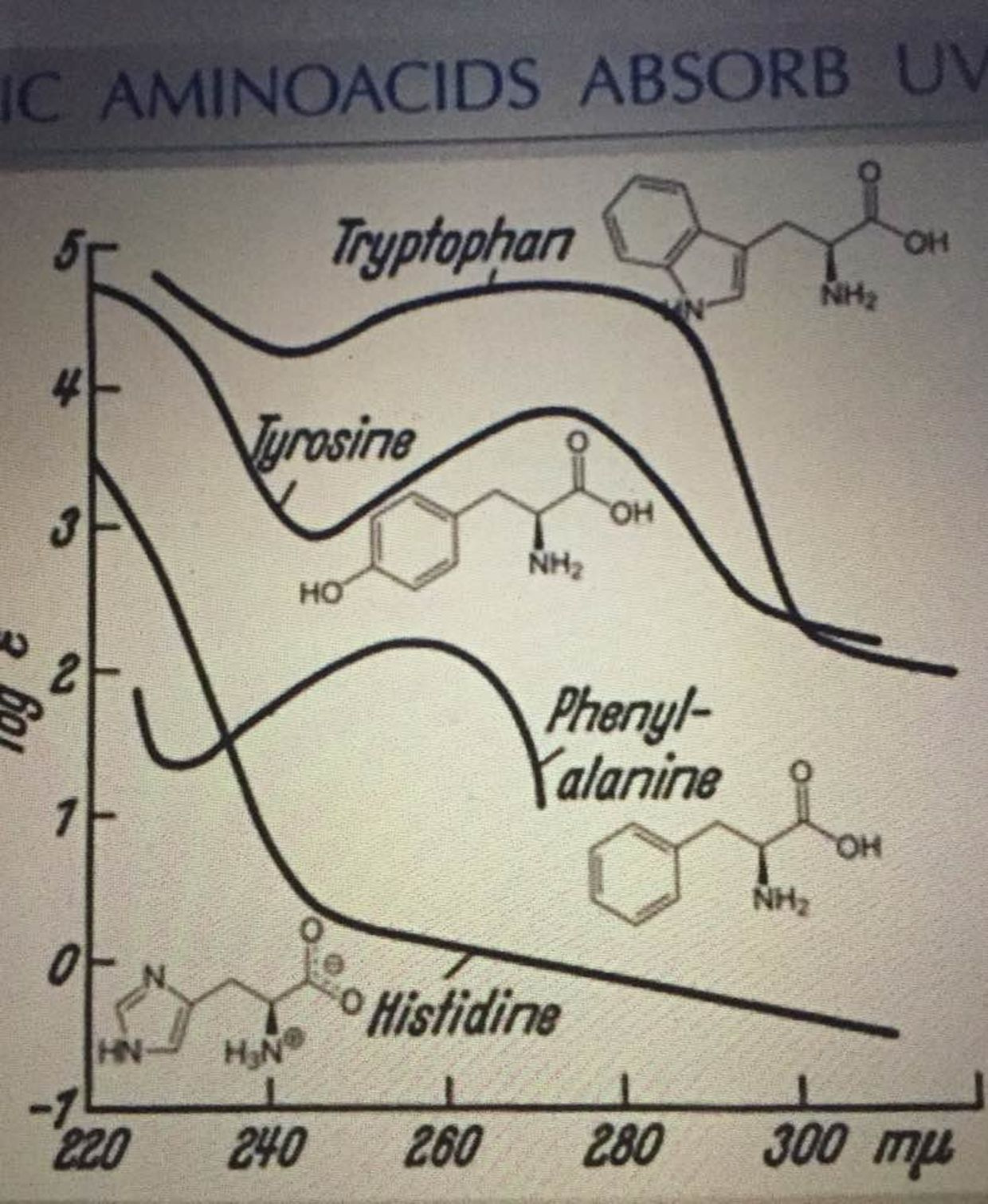
NAD+ is a hydride acceptor from the TCA cycle that forms the reduced dinucleotide NADH. The NAD+/NADH nucleotide pair is vital for driving reduction-oxidation (redox) reactions in energy production to oxygen in human mitochondria. Furthermore, NAD+ is a precursor for the phosphorylated dinucleotide pair NADP+/NADPH, which is required for several cellular biosynthetic pathways (Pentose phosphate pathway =PPP) and to protect cells from reactive oxygen species (ROS) made in the mitochondrial matrix. So when NAD+ is lowered for any reason, the quantum clinician knows that the matrix is making more ROS/RNS than it should be while liberating more ELF-UV as a result.
This raises another question, where does the ELF-UV light come from?
That answer will be covered soon the QT series here and in my Vermont 2018 talk on June 1.
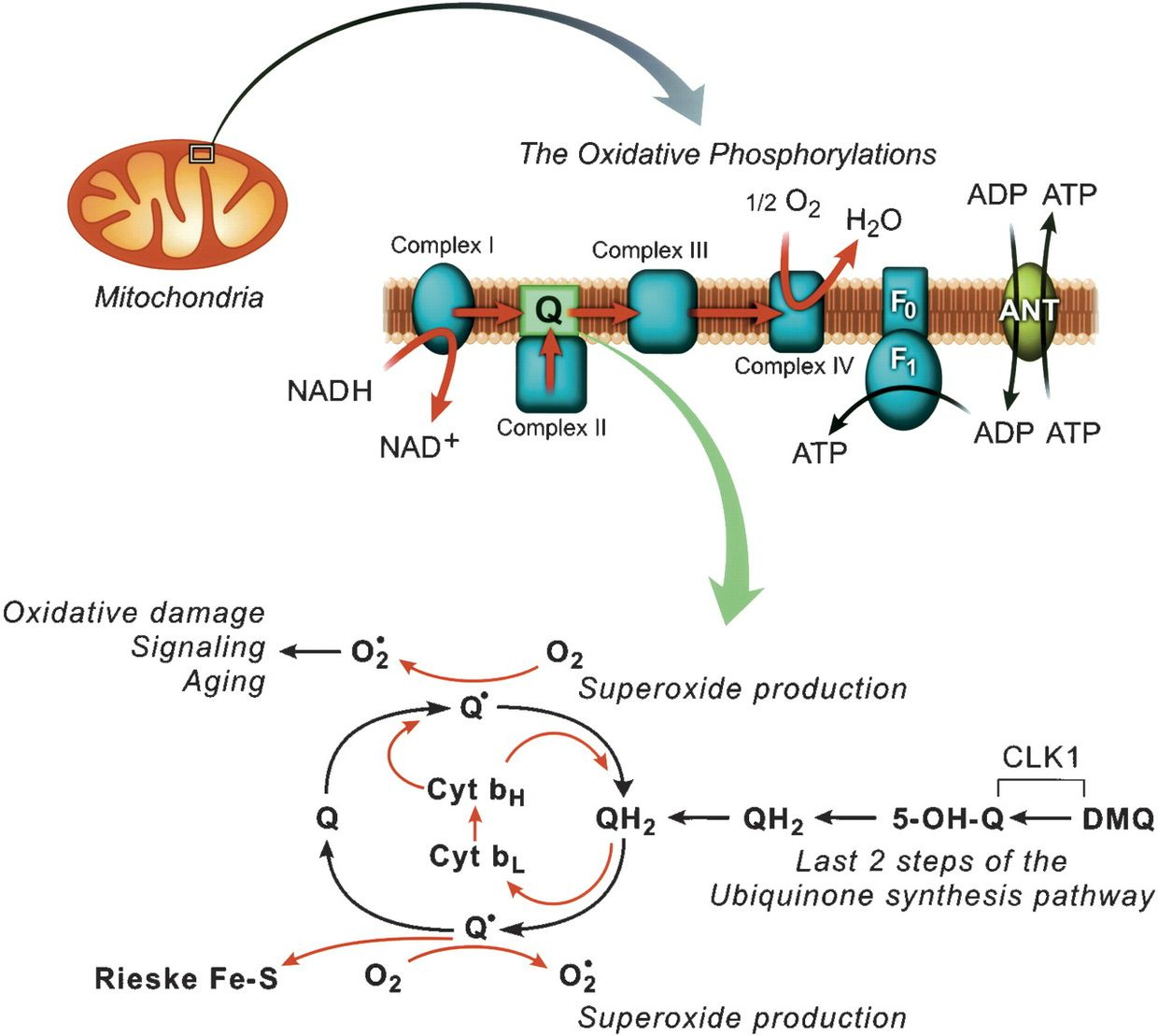
NAD+ is the oxidized version of cytochrome 1 meaning it is lacking an electron from dietary carbohydrate breakdown. NADH is its reduced form of this chemical, meaning it has these electrons from carbohydrates. Together both make up the thermodynamic couple that carries electrons from carbohydrates in humans. NAD+ is part of the cytochrome 1 couple that carries carbohydrate electrons to oxygen in normal mitochondrial function. It is a fluorophore-proteinthat has an absorption spectrum of 340nm which is also deep in the UV range.
KEY BLOG POINT:
The team of researchers at the University of Texas from cite 1 below has found NAD+ synthesis and consumption integrate glucose metabolism and adipogenic transcription during adipocyte differentiation. In their paper published in the journal Science, the group described their research into how glucose is converted into fat in the body. As obesity rates continue to climb around the globe, these scientists wanted to explore why obesity is rising as NAD+/NADH couple in mitochondria is defective. None of them realize that nnEMF cause AMPk amplification and more glucose to be spilled in the blood because cytochrome 1 is destroyed by the frequencies of light used by modern communications. The defect can be enhanced by blue light toxicity in the skin and eye because as melanopsin is ruined, melatonin is lowered and we cannot maintain our mtDNA to keep the cytochromes in ECT working well or replace them. UV sunlight helps us eat less by the design in our mitochondria.

They do not seem to realize that UVA light from the sun has to excite these electrons to get the effect. Just eating tremendous amounts of them does nothing but cause them to go to storage via the NAD+ PPP pathways. They also do not seem to be aware that the H+ need to make NADH whole again must come from the mitochondrial matrix. It cannot be in any other isoform of hydrogen. The May 2018 webinar laid the foundations out for you.

If these light conditions are NOT met on the skin and eye, NAD+ will remain abnormal. This is why blood glucose rises. The electrons cannot be fed in if cytochrome 1 is nonfunctional. As blood glucose rises, insulin secretion increases as a collateral issue from beta cells and this causes the fat mass to expand. The cause of diabetes is really mitochondrial dysfunction and not an insulin driven mechanism. This is why the UT researchers remain confused. They do not realize where the pieces fit.

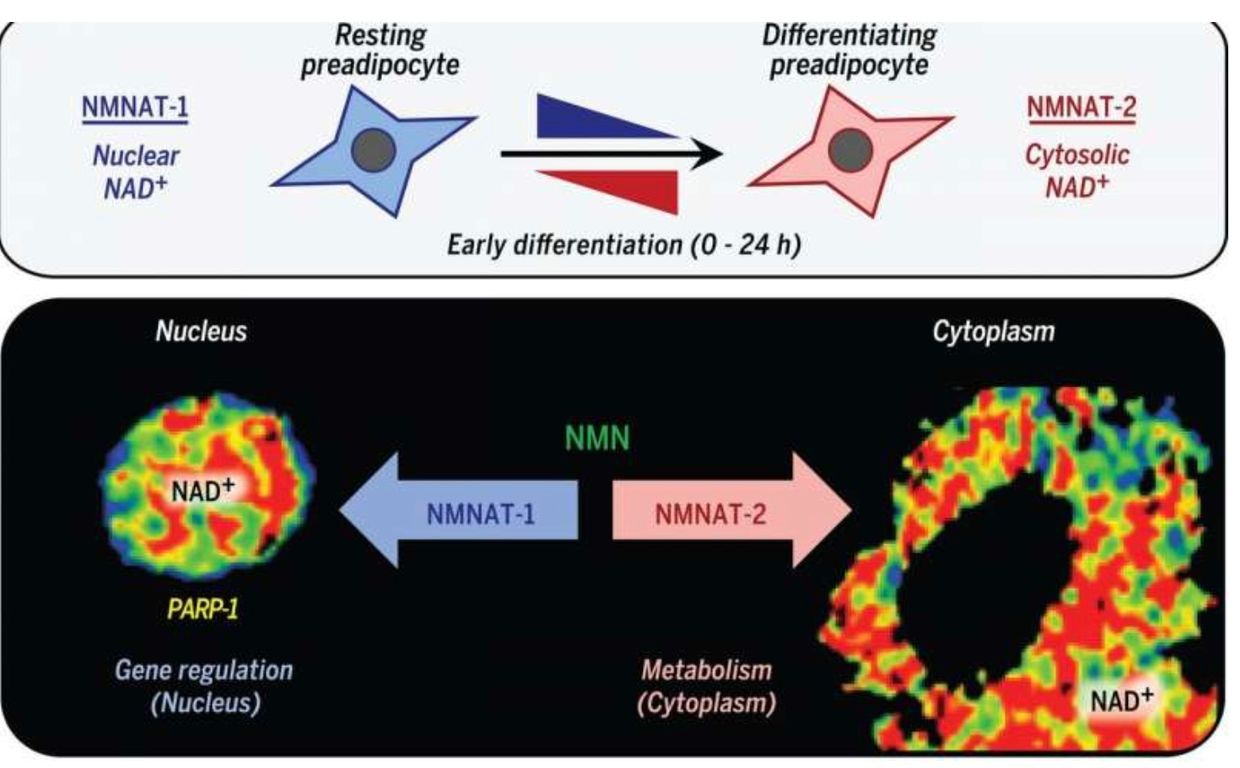
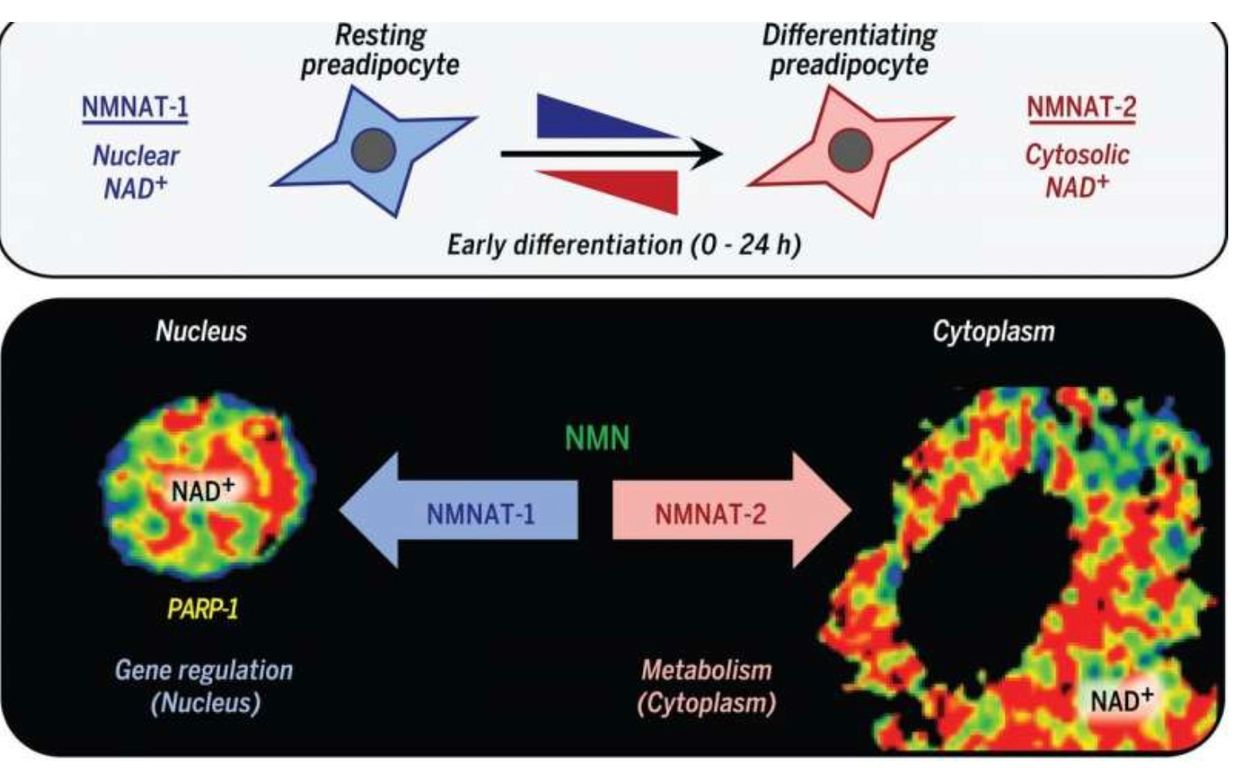
NAD+ synthesis is compartmentalized and within several membrane-bound organelles associated with the outer mitochondrial membrane via the Tensegrity system. It is synthesized in distinct subcellular compartments by three different synthases called NMNAT-1, -2, and -3 as the picture shows.
KETOSIS IS SUPPOSED TO BE USED FOR OUR OWN FAT OR IN WINTER, NOT 24/7.
We are designed to use our OWN fat in the subcutaneous space in autumn and winter to run these programs when UVA and UVB light are absent. Eating fat is not the same idea.
Eating a high-fat diet is one way to raise NAD+, but eating this way poses a risk if the mitochondrial colony in that person’s tissues cannot effectively use beta-oxidation of the TCA cycle. In this case, the nutritional ketosis drives an increased adipogenesis via adipogenic signaling that is rapidly induced by the cytoplasmic NMNAT-2.
This isoform of the synthetase competes with a nuclear version called NMNAT-1 for the common substrate, nicotinamide mononucleotide. This chain of events leads to a precipitous reduction in nuclear NAD+ levels. This signal inhibits the catalytic activity of poly-adenosine diphosphate (ADP)–ribose] polymerase–1 (PARP-1). This is an NAD+-dependent enzyme that represses adipogenic transcription by ADP-ribosylating the adipogenic transcription factor C/EBPβ. So when somebody advocating a high-fat diet in the face of mitochondrial damage due to a chronic non-operational cytochrome one, you can get quite fat from eating fat.

This is especially true when the TCA cycle is dysfunctional for any reason. Blue light toxicity cause breakdown of the outer mitochondrial membrane and this allows deuterium to enter the TCA and urea cycle. This destroys the kinetics of the NAD+/NADH couple. It has many more collateral effects. Most people in the LCHF community have no clue this mechanism exists because they do not read mitochondrial papers carefully. In fact, the reversal of PARP-1–mediated repression by NMNAT-2–mediated nuclear NAD+ depletion occurs in response to adipogenic signals which drives adipogenesis in humans.
This is the dominant way technology use causes obesity via the melanopsin signaling in the eye, skin, and fat. I call this the Jimmy Moore effect. Why?
As NAD+ drops because of its own metabolic consumption, there is a serious loss of information transfer in mitochondria. This is why low NAD+ levels are associated with aging and obesity and many other chronic diseases. If your TCA and urea cycle are broken, oxygen levels also drop. This drop in oxygen means the cell CAN ONLY use glycolysis and the PPP to drive biosynthesis.
IS NAD+ AFFECTED BY THE PPP?
Yes it is.
NAD+ is broken down into nicotinamides and ADP-ribose that feed into the PPP. Glycolysis and the PPP are older metabolic pathways involved in cell biosynthesis that operate in lowered oxygen environments with poorer redox capability. These two pathways also get pulled into double duty when the TCA or urea cycle are non-functional because of the blockade at fumerase. Fumerase is where both of these cycles meet at the cell membrane. This situation is usually caused by cell membrane damage in the mitochondria which unleashes deuterium in the matrix where these two pathways meet one another. These two pathways were favored by the first two kingdoms of life prior to the Cambrian explosion and early after the Cambrian explosion before atmospheric oxygen spiked for mitochondria to take full advantage of the electron negativity to pull electrons with some force toward oxygen. This means that NAD+ must be resynthesized in some fashion FAST enough for a normal cellular function to continue. Many researchers have noted that some prior papers have suggested that lower-than-normal levels of NAD+ can alter metabolism, leading to higher disease susceptibility.

Is there another way to help NAD+ levels when the TCA and urea cycle are functioning poorly? Yes. We can use polyphenols that affect the sirtuin axis. This is why I am fond of high polyphenol wines from high altitudes. They have two beneficial effects on hydrogen for the NAD+/NADH couple. The second way they operate is that these grapes are grown with deuterium depleted rainwater at higher altitudes and latitudes. I have found some coffees can do this as well and will be talking about them in the future. In particular, NAD+ functions as a co-substrate in deacylation reactions which are carried out by the sirtuin family of enzymes. These NAD+-dependent deacylases control several aspects of metabolism and a wealth of data suggests that boosting sirtuin intake may affect the endogenous activity NAD+. The mitochondrial sirtuins control metabolism and information transfer in the matrix by coupling NAD+ consumption with deacylation on critical lysine residues of metabolic proteins in the mitochondria, thereby regulating flux through cellular metabolism This requires the use of some salt and a higher saturated fat diet regimen loaded with seafood and pork. Soon you’ll find out why this is the case here on Patreon.
CITES:
1. Keun Woo Ryu et al. Metabolic regulation of transcription through compartmentalized NAD+biosynthesis, Science (2018). DOI: 10.1126/science.aan5780
2. Achanta L. B., Rae C. D. (2017). β-Hydroxybutyrate in the brain: one molecule, multiple mechanisms. Neurochem. Res. 42, 35–49. 10.1007/s11064-016-2099-2
3. Ahn Y., Narous M., Tobias R., Rho J. M., Mychasiuk R. (2014). The ketogenic diet modifies social and metabolic alterations identified in the prenatal valproic acid model of autism spectrum disorder. Dev. Neurosci. 36, 371–380. 10.1159/000362645
4. Belenky P., Bogan K. L., Brenner C. (2007). NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19. 10.1016/j.tibs.2006.11.006
5. Boison D. (2017). New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 30, 187–192. 10.1097/wco.0000000000000432
6. Bough K. J., Rho J. M. (2007). Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48, 43–58. 10.1111/j.1528-1167.2007.00915.x
7. Bough K. J., Wetherington J., Hassel B., Pare J. F., Gawryluk J. W., Greene J. G., et al. . (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. 10.1002/ana.20899
8. Braak H., Braak E. (1998). Evolution of neuronal changes in the course of Alzheimer’s disease. J. Neural Transm. Suppl. 53, 127–140. 10.1007/978-3-7091-6467-9_11
9. Branco A. F., Ferreira A., Simões R. F., Magalhães-Novais S., Zehowski C., Cope E., et al. . (2016). Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur. J. Clin. Invest. 46, 285–298. 10.1111/eci.12591
10. Brownlow M. L., Benner L., D’Agostino D., Gordon M. N., Morgan D. (2013). Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS One 8:e75713. 10.1371/journal.pone.0075713
11. Brownlow M. L., Jung S. H., Moore R. J., Bechmann N., Jankord R. (2017). Nutritional ketosis affects metabolism and behavior in Sprague-Dawley rats in both control and chronic stress environments. Front. Mol. Neurosci. 10:129. 10.3389/fnmol.2017.00129
12. https://forum.jackkruse.com/index.php?threads/question-reversing-aging-by-restoring-nad-levels.11588/#post-138939