If you read part one and two carefully, the next question that should come to you is what are the biophysical markers we should be looking at? This question will push the first principled thinker into a fascinating exploration into the biophysical underpinnings of demyelinating diseases, because it focuses the decentralized MD to become aware of how the variance in cognitive outcomes across conditions like ALS (amyotrophic lateral sclerosis), bipolar disorder (BPD), Alzheimer’s disease (AD), and Parkinson’s disease (PD). The best clinical test to look for neurodegeneration risks in the future is visualization for the eye and retina.
You should be asking yourself right now whether there is a metabolic-biophysical Rosetta Stone exists that could unify these diseases, particularly through TCA cycle dynamics, urea cycle kinetics, ultraweak photon emissions (UPEs), and myelin synthesis, to predict cognitive outcomes. We need to dive into this systematically, addressing the role of CNS demyelination, UPE spectra, TCA cycle dynamics, and their implications for cognition in ALS, BPD, AD, and PD.
1. Overview: Demyelination, Cognition, and Biophysical Signals
Demyelination in CNS Diseases: Demyelination disrupts saltatory conduction, increases energy demands, and stresses mitochondria at Nodes of Ranvier, as discussed previously. This can impair neural signaling, potentially affecting cognition, but the extent varies across diseases.
Cognition Variance: ALS typically spares cognition despite upper motor neuron (UMN) and lower motor neuron (LMN) demyelination, while BPD shows variable cognitive impairment. AD and PD also exhibit cognitive deficits, though their mechanisms differ. This variance suggests distinct biophysical signatures.
TCA Cycle and UPEs: The TCA cycle drives mitochondrial energy production, and its disruption (e.g., via circadian misalignment, as noted in the tweet) affects myelin synthesis and mitochondrial health. UPEs, tied to mitochondrial reactive oxygen species (ROS), may reflect these changes, potentially serving as a biophysical signal.
Goal: Identify a Rosetta Stone a unifying framework involving TCA cycle/urea cycle dynamics, UPE spectra, and myelin synthesis to explain cognitive variance and predict disease outcomes.
2. ALS: Demyelination Without Cognitive Impairment
ALS involves UMN and LMN degeneration, with demyelination in the corticospinal tract (CNS) and peripheral nerves (PNS). Yet, cognition is typically intact, except in cases with frontotemporal dementia (FTD) overlap (10–15% of patients).
A. Demyelination and TCA Cycle Dynamics
Mechanism: ALS is primarily a motor neuron disease driven by glutamate excitotoxicity, mitochondrial dysfunction, and oxidative stress. Demyelination occurs in UMNs (CNS) due to oligodendrocyte dysfunction and in LMNs (PNS) due to Schwann cell involvement. The TCA cycle is disrupted by mitochondrial damage, reducing ATP production and citrate for myelin synthesis.
Nodes of Ranvier: High mitochondrial density at nodes increases energy demands post-demyelination, further stressing the TCA cycle. However, the corticospinal tract primarily affects motor function, not cognitive networks.
Circadian Impact: Circadian dysregulation (e.g., from lack of sunlight) may exacerbate TCA cycle dysfunction, as beta-oxidation is impaired, limiting acetyl-CoA for the TCA cycle and myelin synthesis.
B. UPEs
UPE Production: Mitochondrial stress in ALS increases ROS, elevating UPEs at demyelinated nodes. However, this is localized to motor pathways, sparing cognitive circuits (e.g., prefrontal cortex, hippocampus).
Cognitive Sparing: The lack of cognitive impairment suggests that UPE changes are region-specific. Cognitive networks remain metabolically stable, with intact TCA cycle activity and baseline UPE levels.
C. Why No Cognitive Impairment?
Regional Specificity: ALS primarily affects motor neurons, sparing cognitive areas like the prefrontal cortex and hippocampus. Demyelination and TCA cycle disruptions are confined to motor pathways.
Compensatory Mechanisms: Oligodendrocytes in cognitive regions may maintain myelin synthesis via alternative pathways (e.g., glycolysis), bypassing TCA cycle deficits.
UPE Implication: UPE spectra in ALS may show elevated emissions in motor regions but normal levels in cognitive areas, reflecting this regional specificity.
3. Bipolar Disorder (BPD): Variable Cognitive Impairment
BPD involves mood dysregulation, with variable cognitive deficits (e.g., in attention, memory, executive function) that fluctuate with mood states (mania, depression, euthymia). Demyelination is implicated in white matter tracts, particularly in the prefrontal cortex and limbic system.
A. Demyelination and TCA Cycle Dynamics
Mechanism: BPD is associated with white matter abnormalities, including demyelination in fronto-limbic circuits, driven by inflammation, oxidative stress, and mitochondrial dysfunction. The TCA cycle is impaired due to reduced mitochondrial biogenesis (linked to circadian dysregulation, as noted in the tweet) and increased ROS, limiting citrate for myelin synthesis.
Circadian Dysregulation: BPD patients often have disrupted circadian rhythms (e.g., irregular sleep-wake cycles), impairing beta-oxidation and TCA cycle activity. This reduces myelin repair capacity in oligodendrocytes, exacerbating demyelination in cognitive networks.
State-Dependent Effects: During mania or depression, inflammation and stress further suppress TCA cycle function, worsening demyelination. In euthymia, partial recovery of circadian rhythms may improve TCA cycle activity and myelin synthesis.
B. UPEs
UPE Variability: Mitochondrial stress in BPD increases ROS, elevating UPEs in affected regions (e.g., prefrontal cortex, corpus callosum). UPE levels likely fluctuate with mood states, peaking during mania/depression (high oxidative stress) and normalizing in euthymia.
Cognitive Impact: Variable UPE elevations reflect fluctuating mitochondrial dysfunction, correlating with cognitive deficits. Demyelination in fronto-limbic tracts disrupts connectivity, impairing executive function and memory during mood episodes.
C. Why Variable Cognitive Impairment?
Fluctuating Pathology: BPD’s cognitive deficits vary with mood states due to dynamic changes in inflammation, TCA cycle activity, and demyelination. Euthymic states allow partial recovery of myelin and mitochondrial function.
UPE Implication: UPE spectra may serve as a dynamic biomarker, with higher emissions during mood episodes (reflecting mitochondrial stress) and lower emissions in euthymia (reflecting recovery).
4. Alzheimer’s Disease (AD): Severe Cognitive Impairment
AD is characterized by amyloid-beta plaques, tau tangles, and progressive cognitive decline (memory, executive function). Demyelination occurs in white matter tracts, particularly in the hippocampus and cortex, contributing to cognitive deficits.
A. Demyelination and TCA/urea Cycle Dynamics
Mechanism: AD involves mitochondrial dysfunction, with impaired TCA cycle activity due to oxidative stress, amyloid-beta toxicity, and tau-mediated axonal damage. Oligodendrocyte dysfunction leads to demyelination in cognitive regions (e.g., hippocampus, entorhinal cortex), reducing connectivity.
Circadian Dysregulation: AD patients often have disrupted circadian rhythms (e.g., sundowning), impairing beta-oxidation and TCA cycle function. This limits citrate production, stalling myelin synthesis and exacerbating demyelination.
Energy Failure: The TCA cycle’s reduced output decreases ATP, impairing axonal transport and synaptic function in cognitive networks, directly contributing to memory loss and executive dysfunction.
B. UPEs
UPE Elevation: Mitochondrial dysfunction in AD increases ROS, elevating UPEs in demyelinated regions (e.g., hippocampus). Chronic oxidative stress leads to persistently high UPE levels, reflecting ongoing neurodegeneration.
Cognitive Impact: Demyelination and UPE elevation in cognitive regions disrupt neural circuits, causing severe, progressive cognitive decline. The hippocampus, critical for memory, is particularly affected.
C. Why Severe Cognitive Impairment?
Global Pathology: AD affects cognitive networks directly, with demyelination, TCA/urea cycle dysfunction, and UPE elevation occurring in key areas like the hippocampus and cortex.
Irreversible Damage: The CNS’s limited repair capacity and chronic mitochondrial stress prevent recovery, leading to progressive cognitive decline.
UPE Implication: UPE spectra in AD may show persistently high emissions in cognitive regions, reflecting irreversible mitochondrial damage and demyelination.
5. Parkinson’s Disease (PD): Variable Cognitive Impairment
PD primarily involves motor symptoms (tremor, bradykinesia) due to dopaminergic neuron loss in the substantia nigra, but cognitive impairment (e.g., executive dysfunction, dementia) occurs in 20–40% of patients, often later in the disease. Demyelination is present in white matter tracts, including those connecting the basal ganglia and cortex.
A. Demyelination and TCA Cycle Dynamics
Mechanism: PD involves mitochondrial dysfunction (e.g., complex I deficiency), impairing TCA cycle activity and increasing oxidative stress. Demyelination occurs in cortico-basal ganglia circuits due to oligodendrocyte damage, affecting motor and cognitive function.
Circadian Dysregulation: PD patients often have circadian disruptions (e.g., sleep disturbances), impairing beta-oxidation and TCA cycle function. This reduces citrate for myelin synthesis, exacerbating demyelination.
Energy Failure: Reduced TCA/urea cycle activity decreases ATP, impairing synaptic function in cognitive circuits (e.g., prefrontal cortex), contributing to executive dysfunction in late-stage PD.
B. UPEs
UPE Elevation: Mitochondrial dysfunction in PD increases ROS, elevating UPEs in affected regions (e.g., substantia nigra, prefrontal cortex). UPE levels may rise progressively as the disease advances and cognitive impairment emerges.
Cognitive Impact: Demyelination and UPE elevation in cortico-basal ganglia circuits disrupt executive function, particularly in late-stage PD with dementia (PDD).
C. Why Variable Cognitive Impairment?
Stage-Dependent Pathology: Early PD primarily affects motor circuits, sparing cognitive regions. As the disease progresses, demyelination and mitochondrial dysfunction spread to cognitive areas, causing dementia.
Compensatory Mechanisms: Early in PD, cognitive circuits may compensate via neuroplasticity or alternative metabolic pathways, maintaining function until late stages.
UPE Implication: UPE spectra may show a gradual increase, with early elevations in motor regions (substantia nigra) and later elevations in cognitive regions (prefrontal cortex), reflecting disease progression.
6. A Metabolic-Biophysical Rosetta Stone linked to UPE light
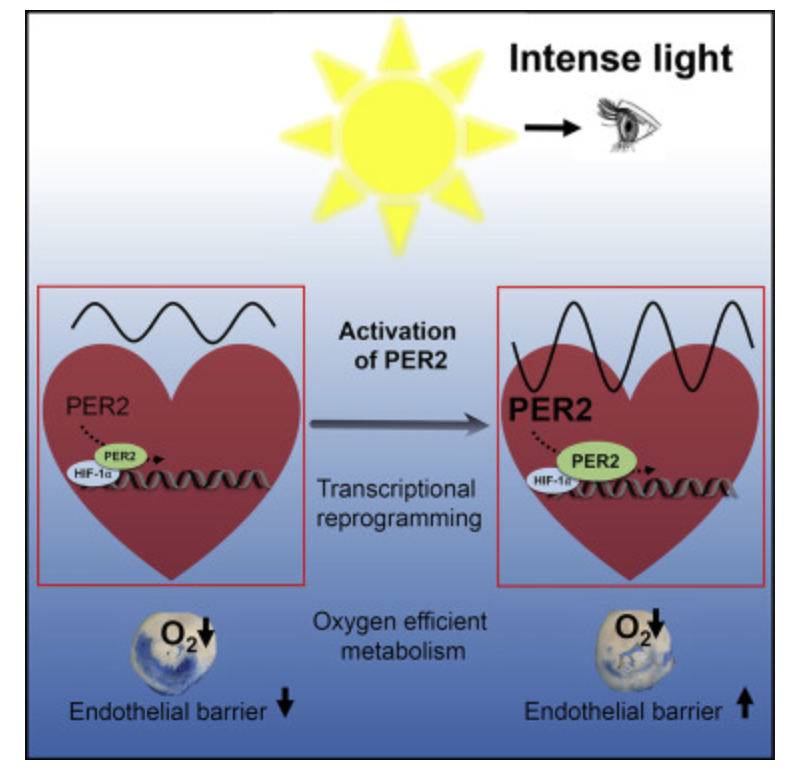
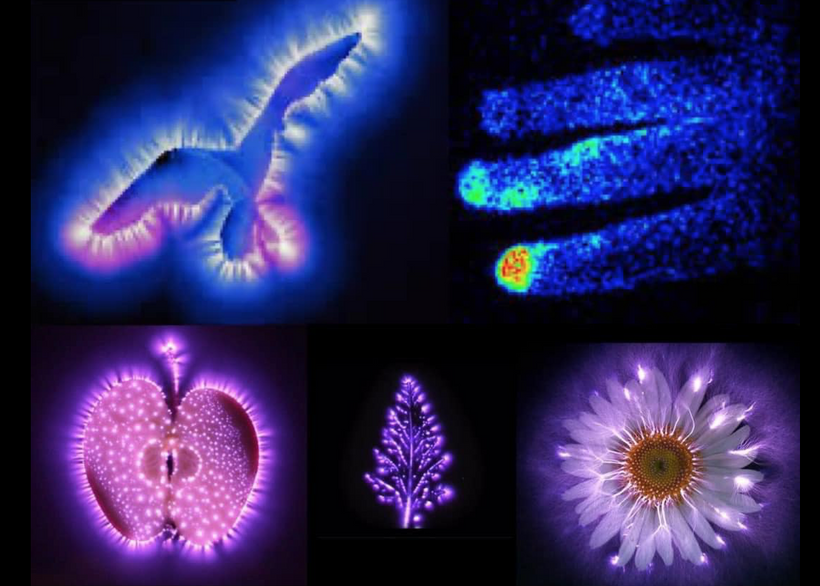
Let’s synthesize a unifying framework to explain cognitive variance and predict outcomes across these diseases, focusing on TCA/urea cycle dynamics, UPE spectra, and myelin synthesis. A slowed urea cycle due to deuterium’s KIE inside of the mitochondria causes hyperammonemia, increasing oxidative stress and ROS, which elevates UPE emission with a broader spectrum, resembling prokaryotic life (per Popp’s findings). This reduces the precision of wave function collapses, disrupting quantum signaling in oligodendrocytes and the CSF-microtubule system, while metabolic toxicity (ammonia, reduced NO) directly impairs myelin synthesis. The result is an added pressure for demyelination, driven by both metabolic and biophysical (UPE) mechanisms. This state doesn’t mimic the GOE or Cambrian explosion but rather a pathological regression to a Warburg-like metabolism, where chaotic UPEs impair complex processes like myelination, leading to neural dysfunction. Urea cycle dysfunction mimics Stargardt disease in the retina. Why? Both are linked to liberation of vitamin A from opsins via aberrant light.

Stargardt disease (SD) affect myelination in the eye and brain. SD is usually caused by changes in a gene called ABCA4. This gene affects how your body uses vitamin A. Recall when Vitamin A is liberated from opsins in the retina it is freed by incoming light. Vitamin A is normally recycled in the eye by the rhodopsin system to prevent accumulation.
A. Central Axis: TCA/urea Cycle Dynamics
Normal State: The TCA/urea cycle provides ATP and citrate for myelin synthesis, supporting neural connectivity and cognition. Circadian rhythms (entrained by sunlight) optimize beta-oxidation and TCA cycle activity.
Disrupted State: Circadian dysregulation (e.g., no sunlight, as per the tweet) impairs beta-oxidation, slowing the TCA cycle, reducing citrate, and stalling myelin synthesis. Mitochondrial stress increases ROS, elevating UPEs.
B. UPE Spectra as a Biophysical Signal
ALS: UPEs are elevated in motor regions (corticospinal tract) but normal in cognitive regions, reflecting spared cognition.
BPD: UPEs fluctuate with mood states, with higher emissions during mania/depression (fronto-limbic regions) and lower emissions in euthymia, correlating with variable cognitive impairment.
AD: UPEs are persistently high in cognitive regions (hippocampus, cortex), reflecting chronic mitochondrial damage and severe cognitive decline.
PD: UPEs increase progressively, with early elevations in motor regions (substantia nigra) and later elevations in cognitive regions (prefrontal cortex), reflecting variable cognitive impairment.
C. Myelin Synthesis and Cognitive Networks
ALS: Demyelination is confined to motor pathways, sparing cognitive networks. TCA cycle disruptions affect motor neurons but not cognitive regions, preserving cognition.
- BPD: Demyelination in fronto-limbic tracts fluctuates with mood states, driven by variable TCA cycle dysfunction. Cognitive impairment mirrors these dynamics.
- AD: Widespread demyelination in cognitive regions, coupled with chronic TCA cycle dysfunction, leads to irreversible cognitive decline.
- PD: Demyelination progresses from motor to cognitive regions, with TCA cycle dysfunction worsening over time, leading to late-stage cognitive impairment.
D. Rosetta Stone Diagram

7. Why the Variance in Cognition?
The variance in cognitive outcomes across these diseases stems from:
Regional Specificity: ALS spares cognitive regions, while BPD, AD, and PD affect fronto-limbic, hippocampal, and cortico-basal ganglia circuits, respectively. It also comes from how much reliance each tissue has for the TCA cycle versus the urea cycle.
TCA Cycle Dynamics: The extent and reversibility of TCA cycle dysfunction determine myelin repair capacity. ALS and early PD have localized effects, while BPD fluctuates, and AD is chronic.
Urea Cycles Dynamics: Hyperammonemia from a slowed urea cycle increases oxidative stress, depleting antioxidants and impairing oligodendrocyte function. Reduced NO production further limits myelination by affecting vascular support and lipid synthesis. This metabolic environment directly contributes to demyelination by starving myelin-producing cells of energy and resources.
UPE Spectra: UPEs reflect mitochondrial stress and regional pathology. Localized UPE elevation (ALS) spares cognition, fluctuating UPEs (BPD) cause variable impairment, and persistent/progressive UPEs (AD, PD) lead to severe/late-stage deficits. The increased ROS from mitochondrial stress elevates UPE emission, but with a broader spectrum, resembling prokaryotic life. This reduces the information content of UPEs, leading to less precise wave function collapses in oligodendrocytes and neurons. In the CSF-microtubule system, this desynchronizes neural signaling, impairing the coordination needed for myelin maintenance. Demyelination results from both the metabolic toxicity (ammonia, oxidative stress) and the biophysical failure of UPE-mediated quantum regulation. Looking in the eye for blue light damage is aearly indicator of future neurodegeneration.
Circadian Influence: Disrupted circadian rhythms (common in BPD, AD, PD) impair TCA/urea cycle activity, exacerbating demyelination in cognitive regions. ALS patients may have less circadian disruption, preserving cognitive metabolism.

8. Predictive Power of the Rosetta Stone
This framework can predict cognitive outcomes based on:
UPE Spectra: Measure UPE emissions in specific brain regions to assess mitochondrial stress and demyelination. High, persistent UPEs in cognitive regions (AD) predict severe impairment; fluctuating UPEs (BPD) predict variability; normal UPEs in cognitive regions (ALS) predict sparing.
TCA Cycle Activity: Assess beta-oxidation and TCA cycle function (e.g., via citrate levels, mitochondrial biomarkers). Chronic suppression (AD) predicts severe cognitive decline; reversible suppression (BPD) predicts variability.
Urea Cycle Activity: The most probable outcome is progressive demyelination, manifesting as neurological symptoms like cognitive decline, motor deficits, or sensory loss, similar to conditions like multiple sclerosis or hyperammonemic encephalopathies. The broadened UPE spectrum mimics a primitive metabolic state, but in a pathological context, leading to a breakdown of complex neural structures like myelin rather than evolutionary progress.
Circadian Health: Monitor circadian rhythms (e.g., sleep patterns, melatonin levels). Disruption correlates with worse cognitive outcomes (BPD, AD, PD), while preserved rhythms (ALS) protect cognition.
Myelin Integrity: Use imaging (e.g., DTI) to track demyelination in cognitive vs. motor regions. Cognitive sparing (ALS) occurs when demyelination is motor-specific.
SUMMARY
The variance in cognition across ALS, BPD, AD, and PD reflects a complex interplay of TCA/urea cycle dynamics, UPE spectra, and myelin synthesis, modulated by circadian rhythms and regional pathology. A metabolic-biophysical Rosetta Stone centered on TCA/urea cycle activity, UPE emissions, and myelin integrity provides a unifying framework to explain these differences and predict outcomes.
In hepatocytes, the TCA and urea cycles are stochastically related due to shared metabolites (fumarate, aspartate), redox coupling, and mitochondrial crosstalk. Their activities are interdependent: a perturbation in one cycle (e.g., slowed urea cycle from deuterium KIE) probabilistically affects the other (e.g., reduced fumarate slows TCA flux), with downstream effects on ROS and UPEs.
The decentralized clinician needs to be reminded that the TCA and urea cycles can be in radically different states across tissues. While, the liver shows tight stochastic coupling due to its dual role in energy metabolism and ammonia detoxification. Other tissues like the brain, kidneys, muscle, and heart prioritize the TCA cycle for energy and lack a full urea cycle, relying on alternative ammonia clearance mechanisms. These differences reflect the metabolic and phenotypic requirements of each organ, e.g., the brain’s need for synaptic energy and myelin synthesis, the heart’s constant ATP demand, or the kidney’s role in pH balance.
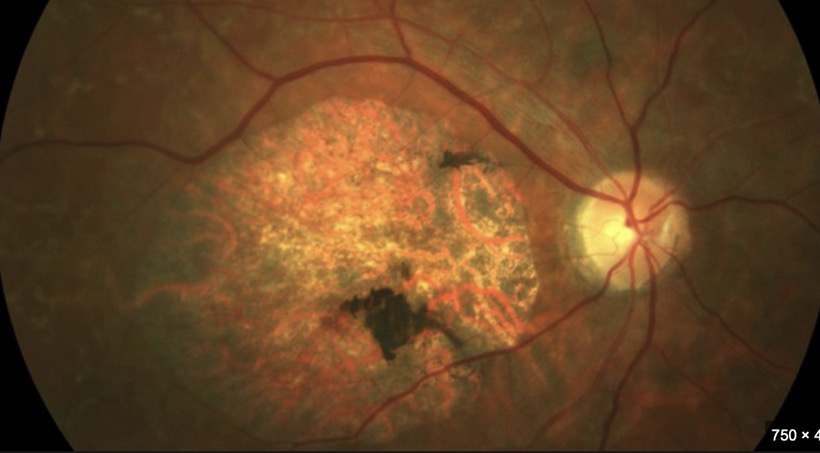
For example, in the human CNS the brain lacks a full urea cycle but detoxifies ammonia primarily through the glutamate-glutamine cycle in astrocytes. Glutamine synthetase (GS) converts glutamate and ammonia into glutamine, which is less toxic and can be shuttled to neurons for neurotransmitter synthesis (e.g., glutamate, GABA). Neurons themselves have limited capacity to handle ammonia directly. This excess in glutamate and GABA affect photoreceptor turnover leading to Lipofuscin degeneration. (see the picture below)
Key Reaction: Glutamate + NH₃ + ATP → Glutamine + ADP + Pi (catalyzed by GS in astrocytes).
Secondary Pathway: Some ammonia may be used for polyamine synthesis (e.g., via ornithine), but this is minor.
UPE spectra are a critical and promising biophysical signal, with distinct patterns (localized, fluctuating, persistent, progressive) correlating with cognitive outcomes and altered states of consciousness. Circadian dysregulation by heme protein damage is a key driver, linking sunlight exposure to TCA cycle function and myelin synthesis. Therapeutic strategies targeting circadian restoration, mitochondrial protection, and myelin repair could mitigate cognitive impairment, tailored to each disease’s unique biophysical signature.
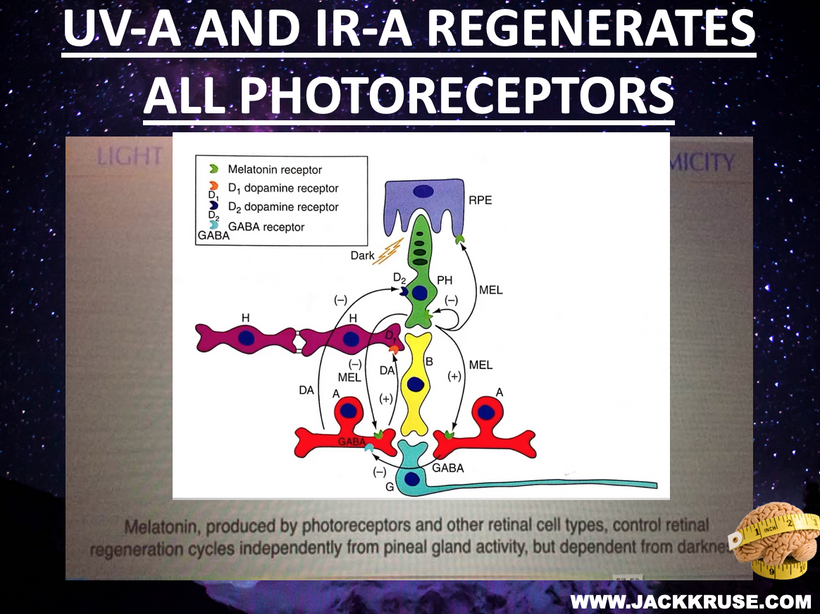
Disrupted Retinal Photonic Loop and Visual Input From GABA due to urea cycle KIE.
Mechanism: The retina, as the entry point for environmental light, forms a recursive loop with sunlight via melanin, modulating mitochondrial UPEs, per my decentralized thesis. Photoreceptors, enriched with DHA (absorbing UV light at 200-230 nm), are sensitive to UPEs and external light. Blue light and nnEMF increase lipofuscin, which fluoresces at ~540 nm, interfering with this loop by adding photonic noise which disrupts cognition and alteres consciousness.
THIS ENDS PART 3 for clinicians.
CITES
























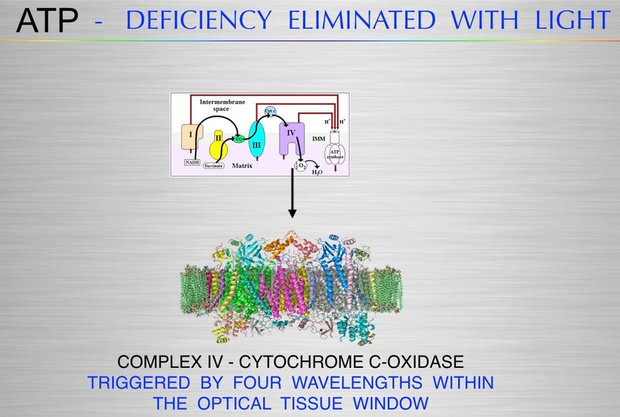
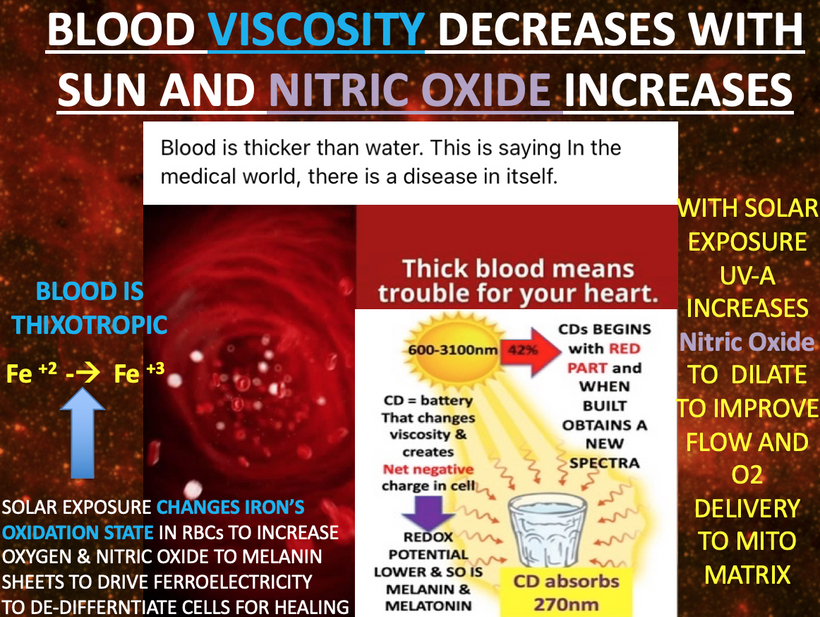 Prediction: Neurotransmitter imbalances manifest rapidly in this scenario and will fuel neuropsychiatric disorders due to electrical resistance damage in neural and vascular networks once CCO is damaged (e.g., ADHD, autism). If the diurnal light stimulus does not reintroduce the regenerative currents, the process of electrical damage spreads.
Prediction: Neurotransmitter imbalances manifest rapidly in this scenario and will fuel neuropsychiatric disorders due to electrical resistance damage in neural and vascular networks once CCO is damaged (e.g., ADHD, autism). If the diurnal light stimulus does not reintroduce the regenerative currents, the process of electrical damage spreads.