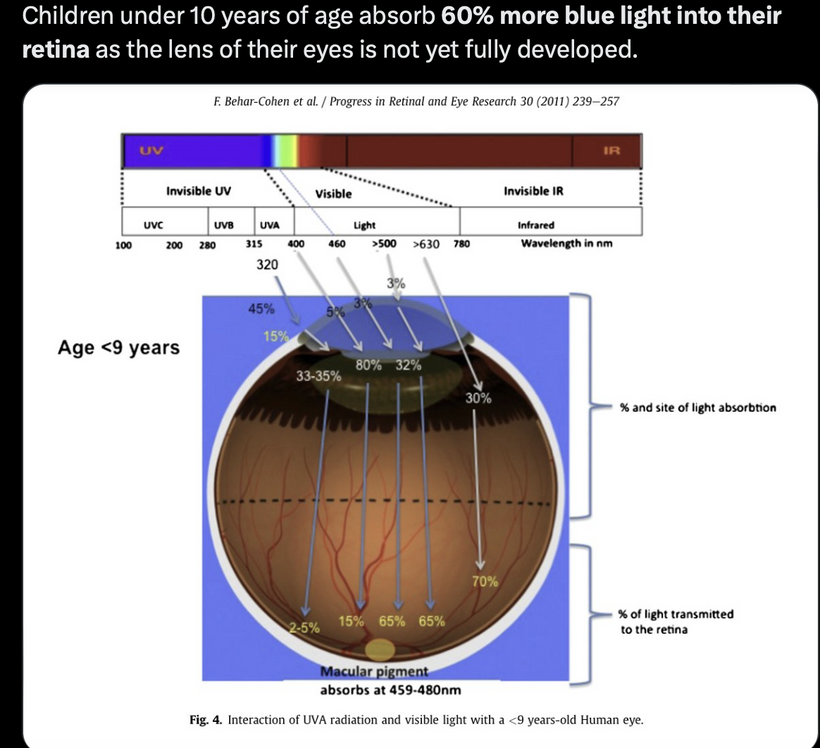
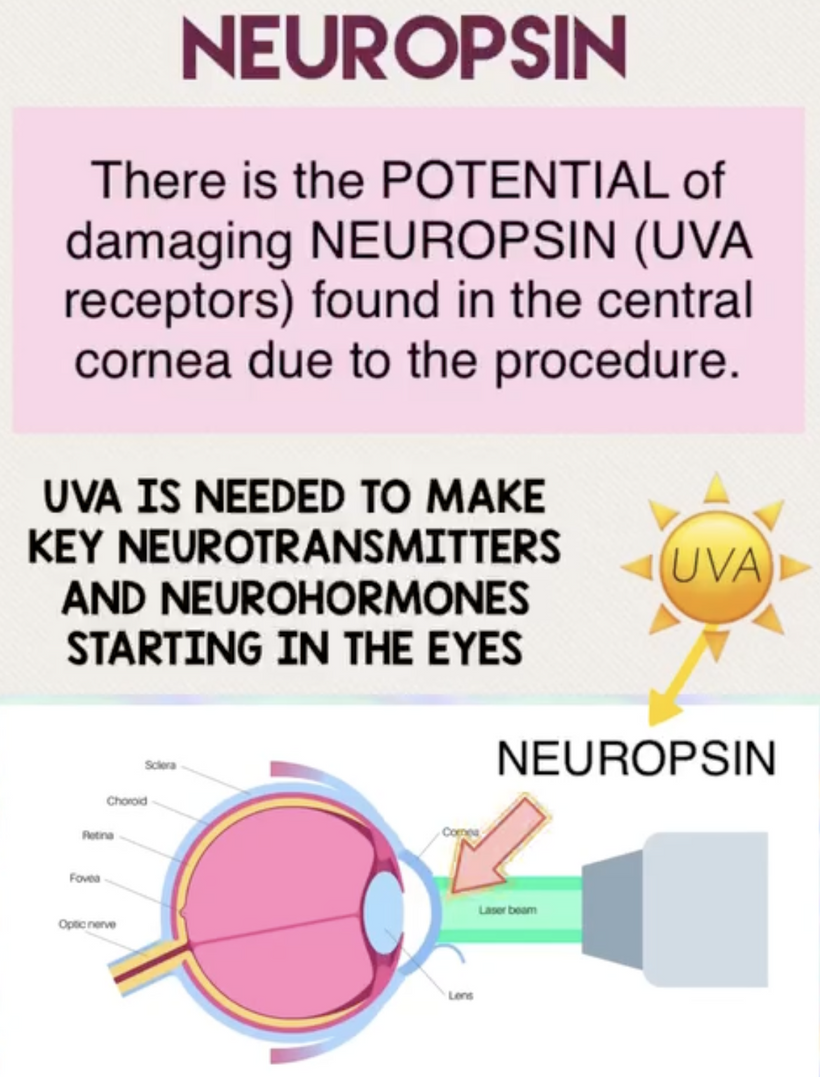
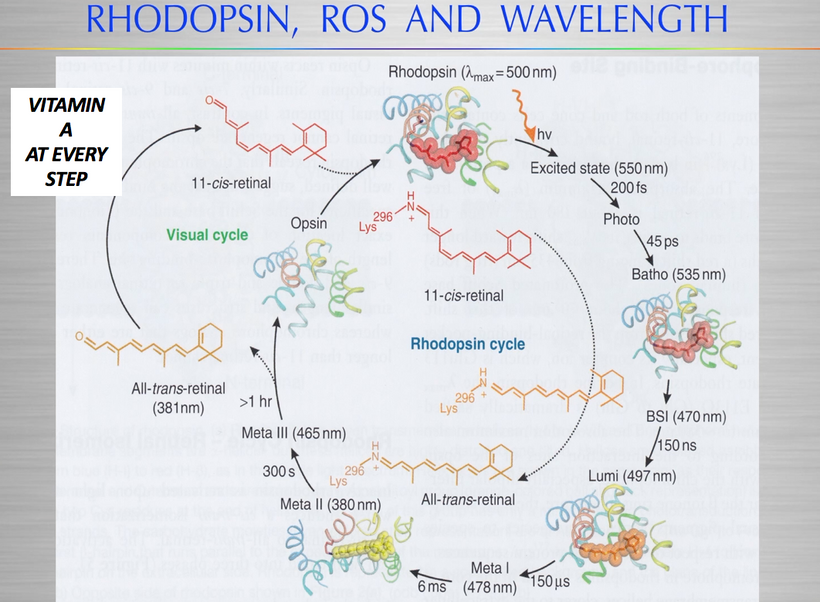
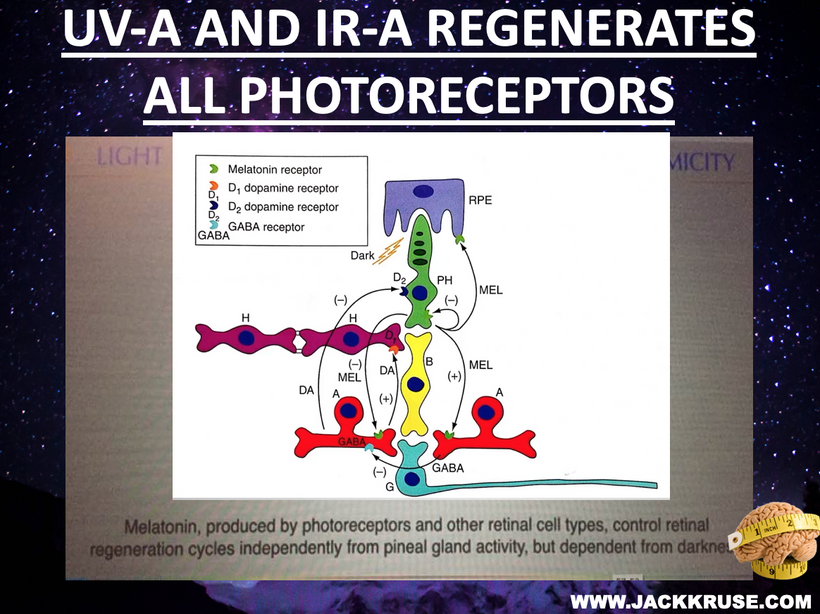
The best cataract avoidance strategy is to stop breaking Nature’s laws around light.
Cataracts, the leading cause of blindness globally (WHO, 2023), not only cause vision loss but also disrupt systemic light-driven processes (e.g., hypothalamic regulation, circadian rhythms). They lead to mitochondrial stress and organ damage (e.g., liver, kidney) due to oxidative imbalance, altered UPE function, and altered mTOR activity. Perception of reality changes as light fails to modulate the distal neural networks in the brain, increasing mental fog, altered consciousness, or misaligned decision-making.
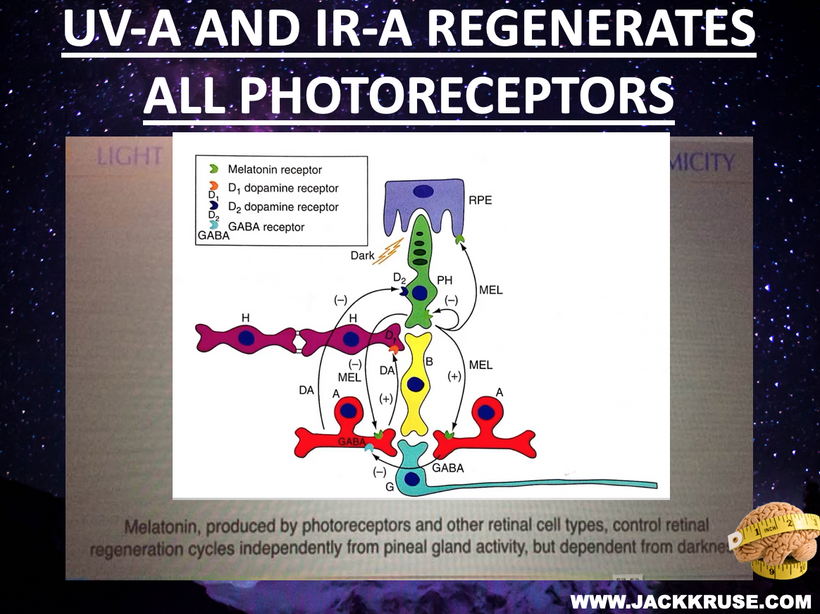
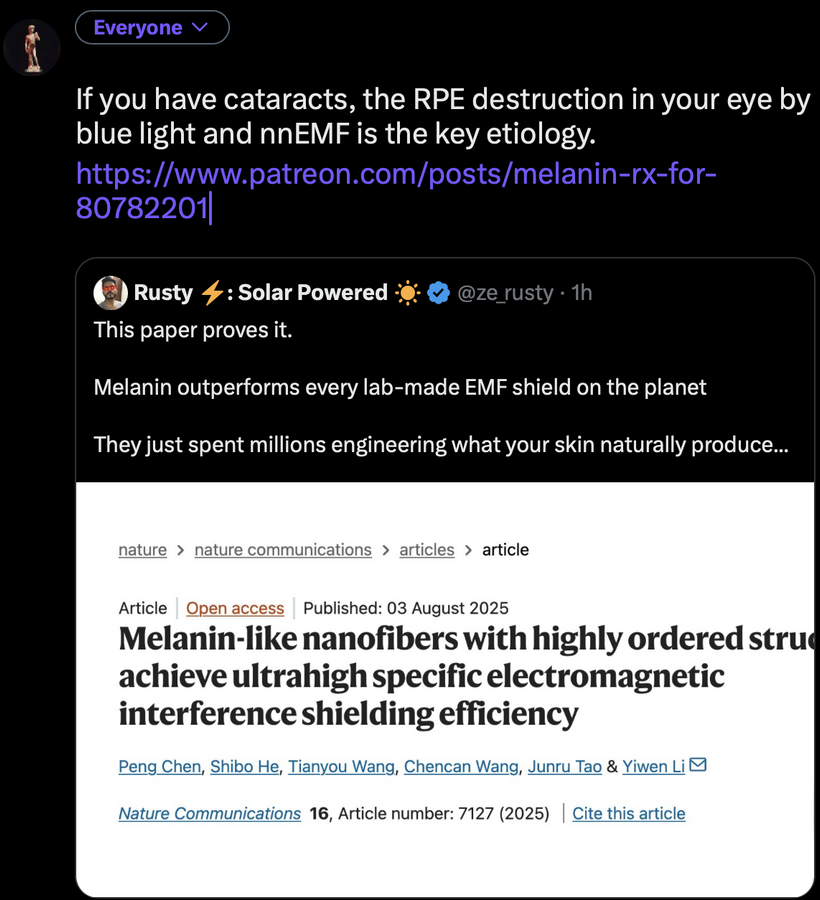
For an audience concerned about cataracts, the information provided highlights a complex interplay between light exposure, melanin, and metabolic processes in the eye. Recent research, such as the August 2025 Nature Communications article, demonstrates that melanin-like nanofibers offer exceptional electromagnetic interference shielding, outperforming lab-made shields. This suggests melanin, naturally produced in the skin and eyes (RPE), serves as a critical defense against non-native electromagnetic fields (nnEMF), which are implicated in cataract formation.
HOW ARE CATARACTS FOUND?
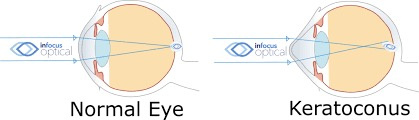
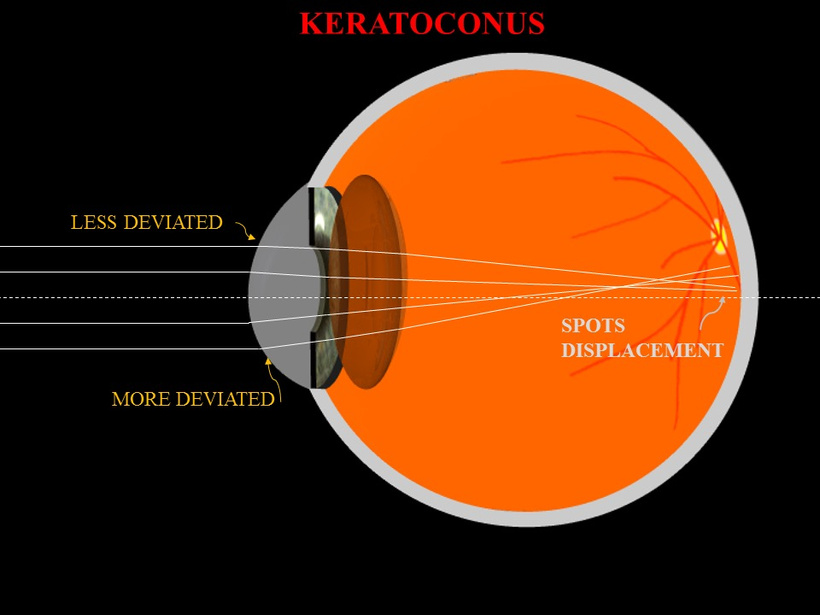
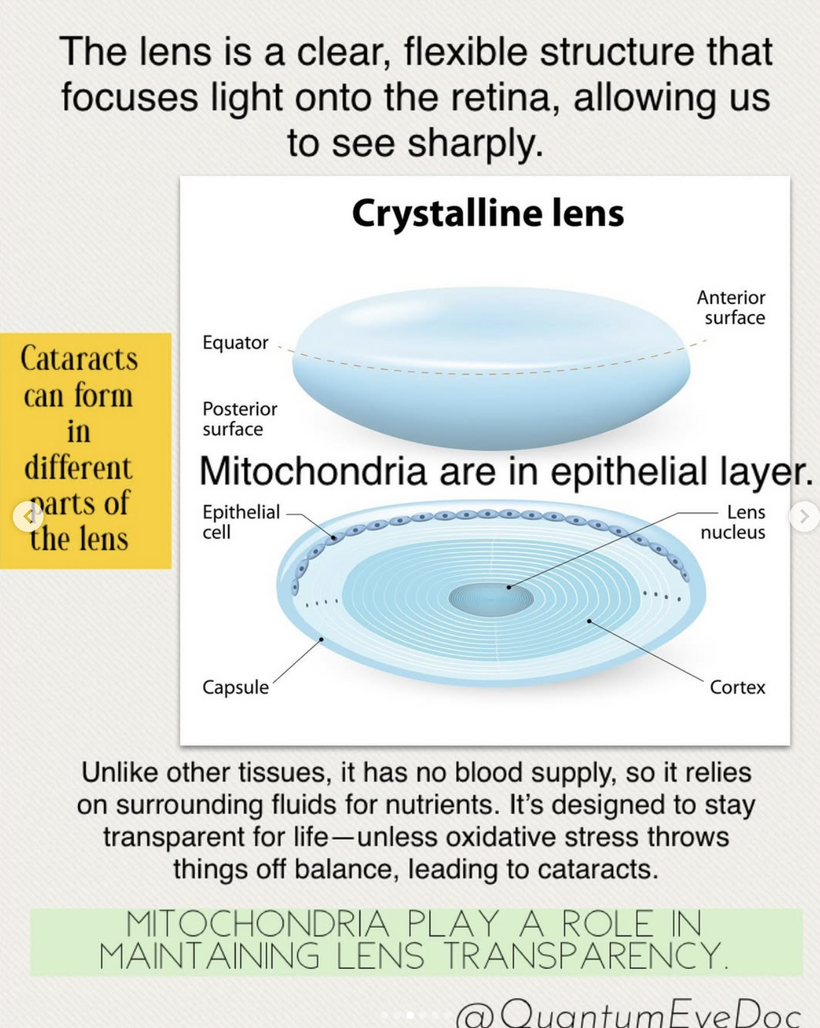
The severity of cataract formation, is primarily assessed using a Snellen visual acuity test. This of course assumes no other ocular disease is present. What really causes the opacification of the lens. The story that follows explains in detail why decentralized medicine > than centralized medicine when it comes to this eye disease.

nnEMF drives high blood glucose and insulin, which causes a hazy lens caused by the brain trying to protect itself. This is the same way tinnitus tries to protect us from acoustic brain damage by putting a melanin sheet between the environment and our acoustic tracts in the brain. No one sees the targets I see in how nnEMF causes these diseases.
The glyoxalase system is a set of enzymes detoxifying methylglyoxal and the other reactive aldehydes produced as a normal part of metabolism. This system has been studied in bacteria and eukaryotes and is linked to human cataract formation.
The glycation of lens proteins essentially drives cataract formation in the eye. The glycation of lens proteins is primarily driven by methylglyoxal. In direct contradiction to much of the low-carbohydrate literature, glycation is not all driven by dietary carbohydrates. It is driven by light. This is why the next slide exists and why it is true.
The 1981 PubMed riboflavin study (https://pubmed.ncbi.nlm.nih.gov/7234715/) links B vitamin deficiencies, which are cofactors in mitochondrial function, leads to redox imbalances in the anterior and posterior eye chambers, contributing to lens opacification. Excessive nnEMF exposure, including blue light and microwaves, disrupts this balance, driving high blood glucose and insulin levels. This metabolic stress leads to glycation of lens proteins, primarily via methylglyoxal, a reactive aldehyde detoxified by the glyoxalase system. Methylglyoxal, is derived from glucose, ketones, or proteins, binds heavy atoms to proteins, impairing semiconduction and causing lens opacity, which are key steps in cataract development.

nnEMF drives opacification of the lens because this enzyme operates with transition metals susceptible to nnEMF in the microwave range. This is why in Dr. Becker’s book, “The Electric Body”, ophthalmologist Savitz found increased cataract formation in people who were exposed to RF/microwaves at their jobs in the 1960s for DARPA. That covers everyone reading this blog today. These cases are easily found with a minimum of research to support Savitz’s work. DARPA destroyed his career because he reported this Becker.


Environments and jobs loaded with this type of nnEMF ruins the heat sink in the eye and distal organs which alters semiconductive current which alter UPEs. When the heat sink is ruined it affects the optics in the lens, eye, and visual tracks. Methylglyoxal is quantitatively the most important source of advanced glycation end products in the body that are linked to the UPE problem. It is not diet that does it.⠀Few people seem to know that, too. The Decentralized Medicine #35 blog on Patreon lays the etiology out for you. But you must read it to gain the wisdom of Nature.

Methylglyoxal can be derived from glucose, or it can be derived from ketones, or it can be derived from protein. So, diet CANNOT be not the driver of this condition; ALAN and a lack of TINA is. The other evidence of proof comes from retinal spacialists themselves. 20 years ago I asked them why does vitrectomy hasten cataract formation if you think the sun causes them? I got a lot of blank stares from them. I explained to them that vitrectomy increases oxygen levels in the eye, which increases ROS and causes more UPEs to be made which damages the anterior and posterior chamber’s redox power. This is the key cause why oxidative damage to the proteins in the lens leading to cataracts. None of them made the link that blue light and nnEMF also raise ROS. I did not expect them to know about Popp and van Wijk’s work on UPEs but they even were ignorant of the basics. This is why they continue to blame diet and sunlight for cataracts. They are a danger to the public health.

This paper below should illuminate how nnEMF destroys glutathione levels via the glyoxalase system. Once you dehydrate melanin sheets of the nerves that innervate the cornea and orbit, you will likely get a cataract. If you are unlucky, you can wind up with a lot worse in your brain due to the electrical scaring. The brain is acting to block blue light from causing this electrical scarring and that is why the lens opacifies. Methylglyoxal causes heavy atoms to bind to proteins they are not supposed to, this ruins semiconduction in the eye and you get an opacified shade on your eye. https://pubs.acs.org/doi/abs/10.1021/jacs.0c01329

Light that comes through the eye programs the hypothalamus. So it stands to reason this evidence that aberrent light would be the leading cause of cataracts, but centralized medicine will produce papers and miss this point, yet, the conclusion of the paper say this: “Conclusions: Our study provides strong evidence that hypothyroidism is a causal determinant of ARC risk”
Lets review what I have taught you alredy in decentralized medicine on this topic.
In a 2025 April blog on hypothyroidism I told you: People with hypothyroidism of any cause will experients sunburning in the sun in a few minutes due to a lack of melanin, melatonin, and Vitamin D. This is due to a lack of Coulomb charge in the skin which alters Gauss law and you cannot collect enough net negative charge in your skin. Well the same thing happens in your lens and this is why it opacifies and ruins your vision. Let’s review why this happens?
Thyrotropic-releasing hormone (TRH) released from neurons in the paraventricular nucleus of the hypothalamus (autonomic center of the brain) is stimulated directly by the hormone, LEPTIN via the leptin-melanocortin pathways.
MASSIVE BLOG POINT: Leptin normally increases melanocortin (α-MSH) and it is required for TRH expression! This means people with hypothyroidism cannot make POMC or melanin well!! This means their longevity will be cut. This is why hypothyroidism is a gateway disease to many others and leads to an earlier demise. Cataracts are one of those gateway diseases. Those with hypothyroidism need massive solar exposure to change their outcomes. This also implies that hypothyroid patients should have worse outcomes from melanin-related diseases, and they DO!
This makes their skin pale, their lens opacify, and these people will also seem to burn more and not know why. Many will develop autoimmune conditions in the skin and gut and their docs will remain impotent to know why. Many will tell you they are allergic to the sun. I just laugh at these comments. When you know better you do better.”
What else did I say in this blog? “TRH serves as a neurotransmitter or neuromodulator outside the hypothalamus. TRH is a general stimulant and induces hyperthermia on intracerebroventricular injection, suggesting a role in central thermoregulation.”
Hyperthermia = heat and heat decreases your heat sink in all things derived from your brain. Your retina is one of those things. When the heat sink is destroyed cataracts become more probable outcome for patients. I also want to remind you that light dark, and temperature control your circadian mechanism. So any form of heat stress is linked to cataract formation. This is called stacking the lessons in the blogs to explain new diseases to you. I mentioned this to several members in the August 2025 Q&A. If you do not stack the lessons you will never learn how the system robs you of time.
Heat intolerance and heat shocks link to many diseases because the lack of water in the heat sink directly alters the UPEs emission spectra you are now learning about. This is why the circadian clock mechanism is linked to the heat shock proteins. Go look at the Quilt document and you’ll notice I talk about them in it.
HIF-1 and HSP90 Interaction
HSP90 is a molecular chaperone that stabilizes client proteins, including HIF-1α, and plays a role in circadian regulation. The interaction between HIF-1 and HSP90 is well-documented in the literature but it is not well known in opthalmology circles.
Stabilization of HIF-1α: Under normoxic conditions, HIF-1α is hydroxylated by prolyl hydroxylases (PHDs) and targeted for degradation via the von Hippel-Lindau (VHL) pathway. HSP90 binds to HIF-1α, preventing its degradation and promoting its stability, even in normoxia. This allows HIF-1α to accumulate and translocate to the nucleus, where it dimerizes with HIF-1β to activate target genes that increase oxygenation and affect TCA metabolism. (e.g., VEGF, EPO).
HSP90 Inhibition: Inhibitors of HSP90 (e.g., geldanamycin) destabilize HIF-1α, reducing its activity. This demonstrates that HSP90 is uber critical for HIF-1 function, particularly in hypoxic or light stressed conditions we see in cataracts.
Circadian Connection: HSP90 also interacts with circadian proteins, including PER2 and CLOCK. It stabilizes these proteins, ensuring proper circadian clock function. For example, HSP90 modulates the nuclear translocation of CLOCK/BMAL1 complexes, which drive PER2 expression. Since HIF-1α and PER2 both rely on HSP90, this chaperone may serve as a molecular hub linking hypoxia and circadian pathways. Why is this big? All these circadian genes are need for PHOTOREPAIR OF CATARACTS. See the picture below for that review. What does this all imply? If your circadian cycle is jacked up, you will NEVER heal your cataracts. This is why eye docs are not taught this biology by the curriculum paid for by BigHarma and intraocular lens companies. They want to sell you drops, potions, and IoL that will make your mtDNA worse so you become a life long patients for other diseases. You clearly have no idea how the centralized Ponzi scheme is built if you are not following this story well enough. How bad are the centralized ideas?
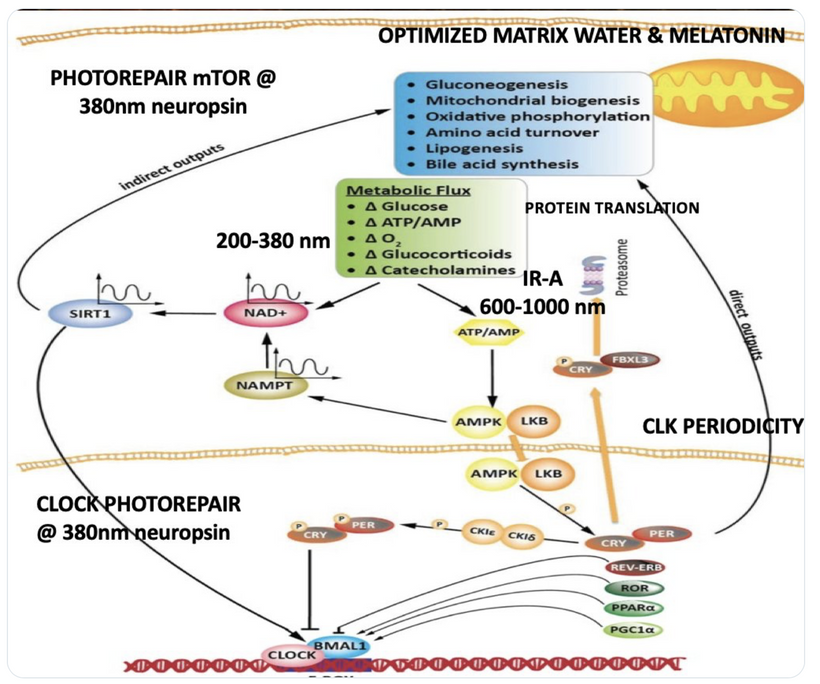
The implications are VAST. Changes in light input through the eye are designed to predict epigenetic programming. What happens when a cataract is between the sun and your DNA, based on my decentralized thesis? Blocked sunlight disrupts histone modifications and DNA methylation, contributing to diseases like cataracts, diabetes, and neurodegeneration, rather than being solely genome-driven. This idea challenges the Darwin-Watson-Crick genomic focus, emphasizing epigenetics (the “junk DNA” or silent genome) is the true regulator of identity and health.
- Circadian and Heat Shock Connections to CATARACTS
Light, dark, and temperature regulate the circadian clock. Hypothyroidism’s impact on melatonin and TRH disrupts this rhythm, exacerbating heat stress and lens damage. Heat intolerance and shocks alter ultraweak photon emissions (UPEs), are linked to circadian and HSP regulation (as noted in the Quilt document). HSPs, stabilizing proteins under stress, tie directly into cataract formation when heat sinks fail in the organs where the disease exists.
Hypothyroidism reduces the lens’s protective mechanisms (melanin, melatonin, vitamin D), impairing its ability to dissipate heat and light. This, combined with TRH-mediated hyperthermia and circadian misalignment, “stacks” stressors that opacify the lens, leading to cataracts. The study conclusion aligns with my thoughts, but its methodology was set up by BigHarma who paid for it so that it would misses the light-heat-cataract nexus I am emphasizing to you in this blog. Wake up. They are lying to you.
MITOCHONDRIA REDOX DAMAGE OPACIFIES YOUR LENS
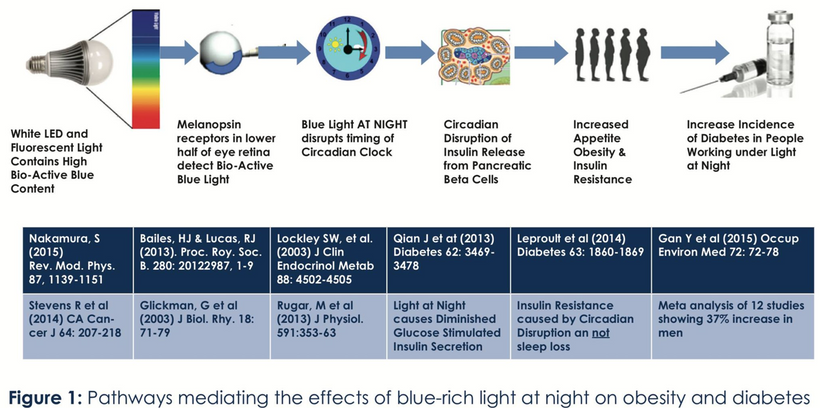
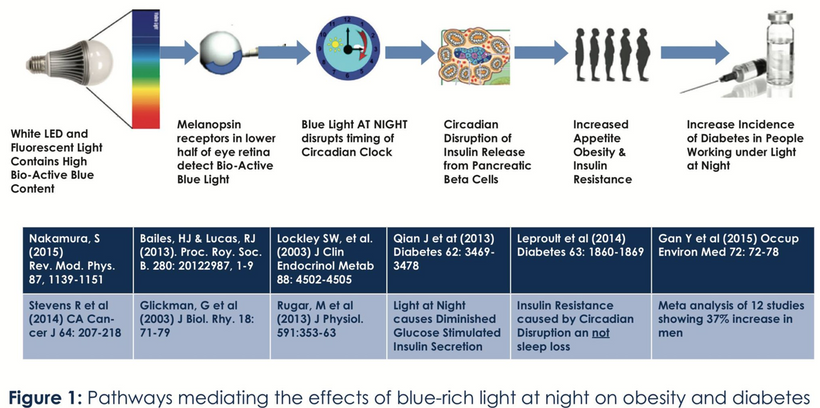
Blue light at night, disrupts the circadian clock, increasing appetite, insulin resistance, and diabetes risk, which further exacerbate lens haze. Dr. Becker’s work, including “The Electric Body” and “Cross Currents,” supports this, noting that heat exposure from nnEMF or blue light ruins the eye’s heat sink (water and melanin-based), linking dry eye syndrome and cataracts.
Dryness, worsened by screen use and reduced hydration, amplifies this effect via Carnot’s theorem, reducing semiconductive efficiency. The best strategy to avoid cataracts is to minimize nnEMF exposure (e.g., reducing blue light at night) and support natural defenses like melanin and glutathione levels. Artificial light at night (ALAN) emerges as a key driver, not just diet, highlighting the need to respect natural light cycles. This multifaceted approach, protecting against nnEMF, optimizing hydration, and maintaining mitochondrial health, offers a proactive defense against cataract formation.

What are the details to this biophysical approach to lens opacification?
THE DESTRUCTION OF THE HEAT SINK OF THE EYE BY BLUE LIGHT and nnEMF
Dry eye syndrome is a common condition in the modern world, with prevalence varying based on screen abuse, climate, ethnicity, lifestyle, and cultural factors. Dry is is related to blue screen technology and as the eye dries, Carnot’s theorem kicks in and the heat sink for semiconduction decreases and this is why dry eye and cataracts are linked. The passage below from the consciousness blogs show you how blue light exposure, poor sleep, and cataracts can begin. The path to getting a cataract is many. There is not one way. Becker showed in Cross Currents that heat exposure alone, from any cause, can lead to cataract formation. He also talked about the work of Dr. Savitz in this disease. Excessive chronic blue light exposure causes dry eye by disrupting hydration signals and vitamin A metabolism. Reduced water production at CCO alters the heat sink (per semiconduction principles below) leading to heat buildup. Heat exposure, as noted by Becker, contributes directly to cataract formation.

What are the neurological loops that control making the eyes wet?
The neurological loops controlling tear production (keeping the eyes wet) involve a complex interplay of the autonomic nervous system, sensory feedback, and central nervous system regulation. Anyone of these pathways can be disrupted to lower tearing to lead to a cataract. Here’s a concise overview of these pathways.
- Lacrimal Functional Unit: Tear production is regulated by the lacrimal gland, accessory glands, and the ocular surface, coordinated via neural pathways.
- Parasympathetic Pathway:
Trigeminal Nerve (CN V): Sensory nerves in the cornea and conjunctiva detect dryness or irritation, sending signals via the ophthalmic branch to the brainstem. This is the nerve of the first pharyngeal arch.
Facial Nerve (CN VII): The superior salivatory nucleus in the pons receives these signals and activates the pterygopalatine ganglion. Postganglionic parasympathetic fibers then stimulate the lacrimal gland to produce reflex tears. This is the second pharyngeal arch innervation.
- Sympathetic Pathway:
The superior cervical ganglion provides sympathetic innervation to the lacrimal gland, modulating basal tear secretion. This is less dominant but helps maintain baseline moisture. This is commonly disrupted in most cases. This pathway also cause dry mouth and lowers parotid salivary flow. This is why many cataract suffers have dry eye and dry mouth and poor dentition.
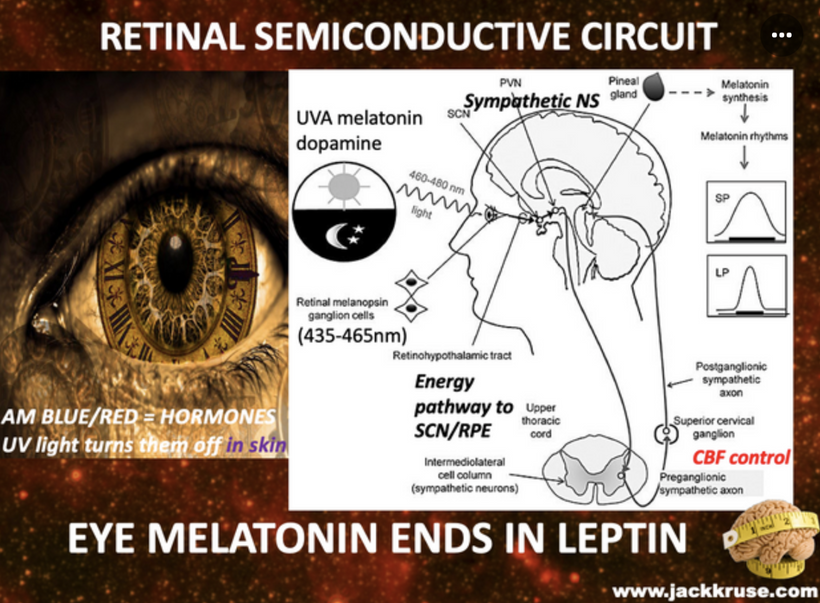
Cataract patients all have mitochondrial inefficiencies. In yesteryear this was blammed on old age, but today nnEMF makes us old when we are young. Patients with low mitochondrial redox frequently experience dry eye and dry mouth, which is linked to autonomic dysfunction, systemic conditions, or medication use. The Nature Communications study of 2025 suggests that sympathetic overactivity in the SCG can suppress lacrimal gland secretion, contributing to aqueous-deficient dry eye, a common complaint in mitochondrial low redox populations.
- Similarly, sympathetic modulation of salivary glands can reduce saliva production, exacerbating xerostomia. Cataracts are often associated with low reodx states like aging, and mitochondrial inefficiencies are more likely to have comorbidities like diabetes, hypertension, or autoimmune diseases (e.g., Sjögren’s syndrome), which can disrupt autonomic innervation. For example, diabetes can cause autonomic neuropathy, affecting both sympathetic and parasympathetic pathways, leading to reduced tear and saliva production. This is why diabetics commonly get cataracts. Diabetes is a blue light toxic disease.
Medications used to manage these conditions, such as beta-blockers or diuretics, are also known to cause xerostomia and dry eye as side effects. This implies they do not treat the proper etiology of the disease, because they do not reverse the dry eye or xerostomia effects. Furthermore, cataract surgery itself can exacerbate dry eye by damaging corneal sensory nerves, reducing reflex tear secretion, and amplifying reliance on basal tear production, which is already be compromised by sympathetic dysregulation. Poor dentition in cataract patients may be a downstream effect of chronic dry mouth, as reduced salivary flow increases susceptibility to caries and periodontal disease. While no single study directly links SCG dysfunction to cataracts, dry eye, dry mouth, and poor dentition, the shared autonomic pathways and systemic factors (aging, medications, comorbidities) provide the quantum biological connection as seen in the picture above.
- Central Regulation:
The hypothalamus and limbic system integrate emotional triggers (e.g., crying) and stress responses, influencing tear production via the autonomic nervous system.
The brainstem (pons and medulla) coordinates reflex tearing in response to environmental stimuli (e.g., wind, , sunlight, and blue light exposure).
- Feedback Loop:
Dryness or blue light-induced disruption affects hydration signals and vitamin A metabolism) reduces sensory input to the trigeminal nerve, dampening the reflex arc. This can lead to decreased tear production, aligning with my heat sink hypothesis. My thesis was the basis of all these predictions in 2010.

WISDOM OF NATURE: A STORY OF BIOPHYSICS
We are all beautiful broken mosaics, pieces of light, love, history, and stars, glued together with quantum magic, music, and words. Do you know where your pieces fit? The glass blower can take shattered glass and apply heat and time to breathe new life into the mosaics, creating an even better version of what existed before. To repair cataracts, you need to understand how the photorepair mechanism in mammals works. This was spoken about in the Quantum Engineering series. This slide is a gentle reminder to re read those blogs.
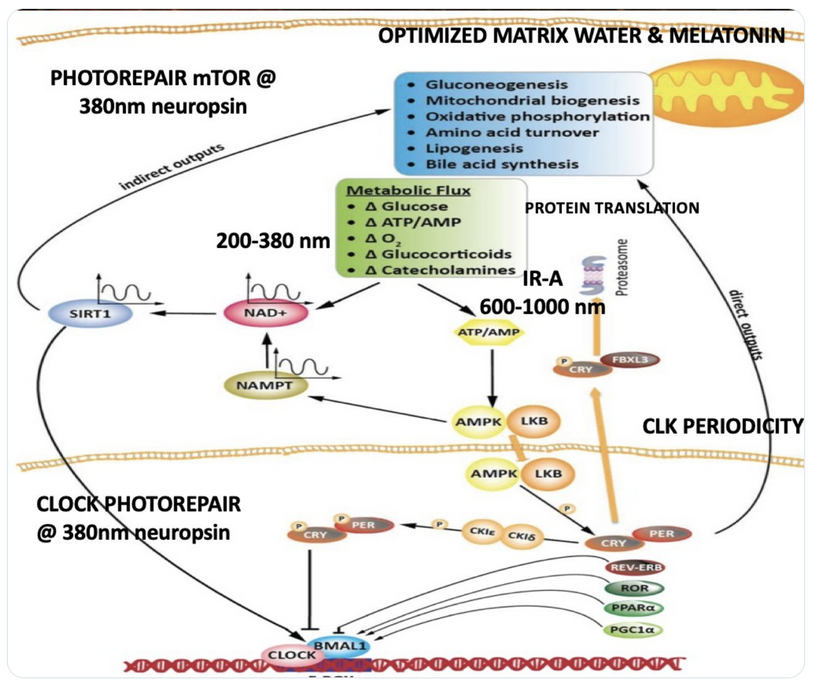
BIOPHYSICS LESSON OF CATARACTS IS A GOE STORY
How much do you know about the decentralized hybrid theory of the Great Oxygen Holocaust in human history? Here is a quick rundown. I know why he got tinnitus, do you? https://threadreaderapp.com/thread/1898906739673563628.html
Modern modeled = a slow evolving zombie apocalypse designed by DARPA. That is how it feels from within when you’re 135 years into the 6th extinction. We like to think we are all smarter than the T-Rexes were, but when you realize we are silly talking monkeys “addicted to our version of the asteroid,” we have no idea what is coming as they did not. I teach Black Swan mitochondriacs how to think to see what few do. Thinking changes your perspective. Many realities are hidden behind a wall of perception. The cause of this extinction is hidden, yet the effect is visible to all mankind.

FIRST PRINCIPLE THINKING ON MY FARM PATIENTS FOR CATARACTS
Considering that we know the innervation of the cornea and orbit as first principle ideas, we should be able to link the lack of melanin to a specific place in the brain to see where nnEMF is causing melanin destruction and mtDNA mutation for water production. For example, we know the human cornea and orbit receive innervation primarily from the ophthalamic branch of the trigeminal nerve (CN V1), with its branches, including the nasociliary nerve and long ciliary nerves, providing sensory innervation to the cornea, and the oculomotor (CN III), trochlear (CN IV), and abducens (CN VI) nerves controlling the extraocular muscles within the orbit. What place in the brain would include where these neural networks connect?
Innervation of the Cornea and Orbit: Neural Pathways
1. Sensory Innervation (Trigeminal Nerve, CN V1):
The cornea is one of the most densely innervated tissues in the body, receiving sensory innervation primarily from the ophthalmic branch of the trigeminal nerve (CN V1).
Nasociliary Nerve: A branch of CN V1, it gives rise to the long ciliary nerves, which innervate the cornea, iris, and ciliary body, carrying sensory (pain, temperature, and mechanical) and sympathetic fibers.
- Pathway to the Brain:
Sensory fibers from the cornea travel through the long ciliary nerves to the nasociliary and ophthalmic nerve (CN V1).
These fibers enter the brainstem at the pons level via the trigeminal nerve root.
They synapse in the trigeminal sensory nucleus, a complex structure spanning the brainstem:
Spinal trigeminal nucleus (caudal part, extending into the medulla and upper cervical spinal cord): Primarily processes pain and temperature sensations from the cornea.
Principal sensory nucleus (in the pons): Processes touch and pressure sensations.
Second-order neurons from the trigeminal sensory nucleus cross the midline and ascend via the trigeminal lemniscus to the thalamus’s ventral posteromedial nucleus (VPM).
From the VPM, third-order neurons project to the primary somatosensory cortex (S1) in the postcentral gyrus (Brodmann areas 3, 1, 2) for conscious perception of corneal sensation.
Associated Areas: The trigeminal system also projects to the insula and anterior cingulate cortex for emotional and autonomic responses to corneal pain (e.g., tearing, blinking). Many cataract sufferers also have comorbid ADHD and OCD. Now you know why.
2. Motor Innervation (CN III, CN IV, CN VI):
The extraocular muscles within the orbit are controlled by:
Oculomotor nerve (CN III): Innervates the superior, medial, and inferior rectus muscles, inferior oblique, and levator palpebrae superioris (for eye movement and eyelid elevation).
Trochlear nerve (CN IV): Innervates the superior oblique muscle.
Abducens nerve (CN VI): Innervates the lateral rectus muscle.
Pathway to the Brain:
CN III (Oculomotor):
Originates from the oculomotor nucleus in the midbrain (at the level of the superior colliculus), which controls voluntary eye movements.
- This also includes the Edinger-Westphal nucleus (midbrain), which is responsible for the parasympathetic innervation of the pupil (via the ciliary ganglion).
Receives input from the superior colliculus (for reflexive eye movements), frontal eye fields (FEF) (Brodmann area 8, for voluntary saccades), and pretectal area (for pupil reflexes).
CN IV (Trochlear):
Originates from the trochlear nucleus in the midbrain (at the level of the inferior colliculus).
Receives similar cortical and subcortical inputs as CN III for coordinated eye movement.
CN VI (Abducens):
Originates from the abducens nucleus in the pons.
Coordinates with the oculomotor nucleus via the medial longitudinal fasciculus (MLF) for conjugate eye movements (e.g., horizontal gaze).
Receives input from the paramedian pontine reticular formation (PPRF) (for horizontal saccades) and the superior colliculus.
Higher-Level Control:
The frontal cortex’s frontal eye fields (FEF) and supplementary eye fields (SEF) plan voluntary eye movements.
The parietal eye fields (PEF) in the posterior parietal cortex (e.g., lateral intraparietal area, LIP) integrate sensory and motor information for spatial attention and eye movement.
The cerebellum (especially the flocculus and vermis) fine-tunes eye movements and coordinates with the brainstem nuclei.
3. Autonomic Innervation (Sympathetic and Parasympathetic):
Sympathetic: The long ciliary nerves (via CN V1) carry sympathetic fibers from the superior cervical ganglion to the dilator pupillae muscle (pupil dilation).
Parasympathetic: The oculomotor nerve (CN III) carries parasympathetic fibers from the Edinger-Westphal nucleus to the ciliary ganglion, innervating the sphincter pupillae (pupil constriction) and ciliary muscle (accommodation).
These autonomic pathways connect to the hypothalamus (for circadian regulation) and pretectal area (for light reflexes).
Where Do These Neural Networks Converge in the Brain?
The innervation of the cornea and orbit involves sensory, motor, and autonomic pathways, which converge in several key brain regions:
Brainstem:
Trigeminal Sensory Nucleus (Pons and Medulla): The first relay for corneal sensory input (pain, touch) via CN V1.
Oculomotor, Trochlear, and Abducens Nuclei (Midbrain and Pons): These control the motor of extraocular muscles, coordinated via the MLF.
Reticular Formation (PPRF): Coordinates conjugate eye movements with input from higher centers. Blue light here is why people with cataracts struggle with sleep. If you have a sleep issue and cataract I know how deep your electrical scar goes in you compared to a newer cataract.
Thalamus: The Sensory Processing behemoth of humans
Ventral Posteromedial Nucleus (VPM): This relays sensory information from the trigeminal system to the neocortex. The less myelin present here from blue light damage the more likley you are to sneeze with bright light exposures.
Lateral Geniculate Nucleus (LGN): Though primarily visual, it integrates with eye movement and pupil control pathways via the pretectal area. This is where many transgeneration causes of eye motions come from via the germline. You heard about these in the August 2025 Q&A when we mentioned amblyopia and nystagmus. Re Listen to it with this blog in mind to see what you missed.
Cortex:
Primary Somatosensory Cortex (S1): Processes corneal sensation (pain, touch).
Frontal Eye Fields (FEF) and Supplementary Eye Fields (SEF): Plan voluntary eye movements. This is why many people have amblyopia and are more prone to seizures from light.
Posterior Parietal Cortex (PPC): Integrates sensory and motor data for spatial awareness and eye movement. This is why so many cataract suffers need Waze and google maps. Their electrical scar in their brain extends here from their cataract defect.
Insula and Anterior Cingulate Cortex: Handle emotional and autonomic responses to corneal stimuli. Many people with cataracts are highly emotional. When the electrical scar is here I look at brain MRIs to see if there is a white matter volume deficit present.
Hypothalamus and Pretectal Area:
Hypothalamus: Regulates autonomic responses (e.g., pupil dilation) and circadian rhythms, influencing melanin production via melanopsin signaling.
Pretectal Area: Mediates pupil light reflexes, connecting to the Edinger-Westphal nucleus.
Cerebellum:
Fine-tunes eye movements and integrates sensory-motor feedback, mainly via the flocculus and vermis. Many people with posterior cataracts get bouts of nystagamus and this is why it can happen. Many people also develop balance problems with cataracts. This is why it happens. This tells the decentralized clinician just how widespread the blue light induced electrical scar is on the neocortex. This directly impacts how long a photorepiar reversal should take.
Linking Lack of Melanin to a Specific Brain Region
The decentralized MD focus should be melanin and its neural crest migratory path. Why? Melanin’s role in bioelectric signaling suggests that a lack of melanin in the cornea and orbit might disrupt these neural networks, particularly at points where melanin-containing cells (e.g., melanocytes) or melanin-dependent bioelectric processes are critical. NCC migration is the motherboard of the cell and explains to us the extent of the electrical resistance in the brain from blue light and nnEMF abuses. Let’s explore this:
Melanin in the Cornea and Orbit:
The cornea itself lacks melanin, but the surrounding structures (e.g., iris, ciliary body) contain melanocytes. The retina (closely related to the orbit) is rich in melanin (e.g., retinal pigment epithelium, RPE). The optic nerve and its projections also involve melanin-containing cells.
As I’ve noted in many blogs, melanin in these areas acts as a photo-bioelectric charge regulator, dampening currents when hydrated and becoming conductive when dehydrated (per my slides and the Popular Science article on eumelanin’s conductivity).
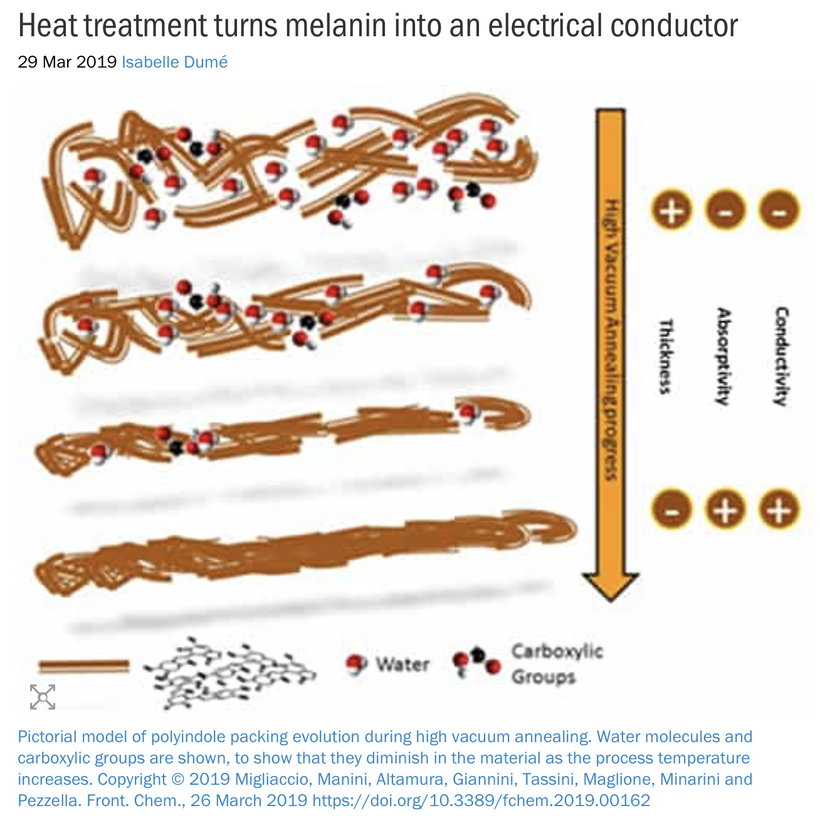
The Impact of endogenous Melanin Deficits in Humans:
Lack of endogenous melanin = destroyed endogenous heat sink = alien UPEs
A lack of melanin (or dehydrated, conductive melanin due to nnEMF/ALAN) in the iris, ciliary body, or retina could disrupt local bioelectric signaling, affecting the trigeminal sensory fibers (CN V1) and autonomic innervation (via CN III). This leads to aberrant signals (e.g., pain, photophobia) or impaired pupil responses, contributing to cataracts or other dysfunctions. This is why TBI patients have photophobia and never show up on imaging. It also explains why concussion patients are at a higher risk to develop cataracts in the future.
In the orbit, extraocular muscles and their innervation (CN III, IV, VI) rely on precise bioelectric coordination. Disrupted melanin in nearby structures (e.g., optic nerve sheath) would alter these signals, affecting eye movement or gaze stability. This is why TBI patients get double vision. Cataracts and TBI share etiology. They both cause organic brain damage.
Central Brain Region Most Affected:
The hypothalamus stands out as a critical convergence point where a lack of melanin has a profound impact:
Melanopsin Connection: The hypothalamus, particularly the suprachiasmatic nucleus (SCN), receives input from melanopsin-containing retinal ganglion cells (via the retinohypothalamic tract). Melanopsin, a light-sensitive pigment, regulates circadian rhythms and pupil responses. A lack of melanin in the retina or iris (due to dehydration or degradation) should impair melanopsin signaling, disrupting hypothalamic control of autonomic functions (e.g., pupil dilation, tear production) and circadian alignment. This is why dry eyes are always linked to cataract formation.
Autonomic Dysregulation: The hypothalamus integrates autonomic inputs from the orbit (via CN III’s parasympathetic fibers and sympathetic pathways). Disrupted bioelectric signaling due to melanin deficits might lead to autonomic imbalance, exacerbating conditions like cataracts & tinnitus (via methylglyoxal accumulation) via downstream effects on auditory pathways in the medial geniculate nucleus).
Photo-bioelectric disruption: My decentralized model emphasizes that dehydrated melanin increases conductivity, amplifying aberrant currents to lead to distal electrical scarring and loss of myelin and MT function. In the hypothalamus, this would disrupt the bioelectric environment, affecting its role in regulating the trigeminal-autonomic reflex (e.g., tearing, blinking in response to corneal irritation) and circadian-driven melanin production. It can also cause memory loss. All of these are seen in TBI patients and most cataract patients.
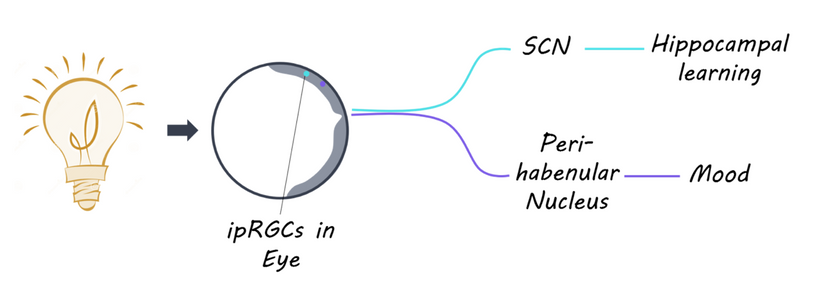
Secondary Impact: Brainstem and Thalamus:
The trigeminal sensory nucleus in the brainstem might also be affected, as it processes corneal sensory input. If melanin deficits in the cornea/orbit lead to aberrant bioelectric signals (e.g., due to nnEMF-induced dehydration), the trigeminal nucleus might overcompensate, contributing to pain or reflex issues. Trigeminal neuralgia could be a photo-bioelectric problem, too. So might intraocular opthalmoplegia which is most often linked to MS. Internuclear ophthalmoplegia (INO) is a disorder of eye movements caused by a lesion in an area of the brain called the medial longitudinal fasciculus (MLF). The most common causes of INO are multiple sclerosis and brainstem infarction. Both of these diseases are also photo-bio-electric diseases associated with demyelination and loss of white matter volumes. In the eyes, OCT studies help predict future risks.
The thalamus (VPM) can misinterpret these signals as a sensory relay, leading to altered perception or chronic conditions (e.g., photophobia, linked to cataracts). TBI patients should wake up about now. I think I just explained symptoms that centralized experts do not. In my worldview, cataracts are brain TBI caused by blue light and nnEMF at chronic levels. The severity of the opacity of the lens links directly to mitochondrial CCO function and VDR on the IMM to protect the mitochondria from stray electrical currents from migrating in the brain. The more UV and IR is subtracted from your life, the worse the opacification will be and the more damage in the brain their will be.

KEY LESSON IN THIS BLOG: Many people do not realize the outer retina has a massive density of mitochondria in the eye and this is why cataracts are so common in our modern world.

Tying these lesson back to My Photo-Bioelectric Framework
My decentralized medicine thesis links nnEMF and ALAN to melanin dehydration, mitochondrial dysfunction, and bioelectric disruption. In the context of the cornea and orbit:
Melanin Dehydration in the Hypothalamus: The hypothalamus, via its role in melanopsin signaling and autonomic control, is a likely central target. A lack of melanin (or its functional degradation) in the retina/iris could impair light-dependent hypothalamic functions, disrupting circadian rhythms and autonomic responses, which in turn exacerbate corneal/orbital pathology (e.g., cataracts via methylglyoxal).
I believe glaucoma is also an nnEMF blue light disease. Why?
My Glaucoma Prediction: Chronic exposure to nnEMF (microwave range) and blue light (e.g., from LED screens) damages melanopsin in retinal ganglion cells (RGCs) and mtDNA in the retinal pigment epithelium (RPE) and optic nerve head. In glaucoma cases patients always show evidence of light damage. They are missing melatonin = lack of UV and IR light. They have low vitamin D levels = lack of UVB exposure and presence of CCO dehydration, and a serious lack of NO = lack of UV and IR light = higher BPs, higher eye pressures, evidence of photoreceptor damage on eye exam.
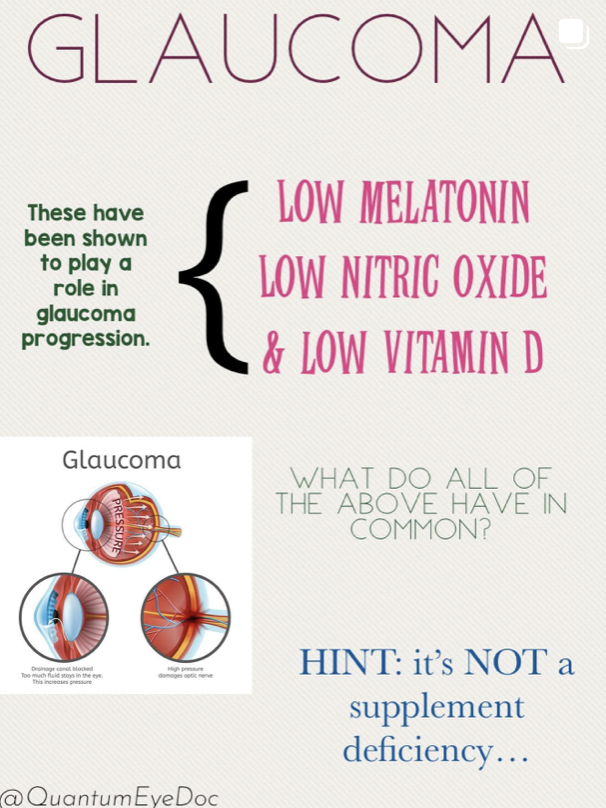
Mechanism: Blue light, absorbed by melanopsin, disrupts the retinohypothalamic tract, altering hypothalamic regulation of circadian rhythms and autonomic tone in the eye. nnEMF, affecting transition metals in the glyoxalase system, depletes glutathione, melanin, and melatonin. These all degrade and lead to abnormal UPE fpormation and this light destroys internal tissue optics. This increasing methylglyoxal and advanced glycation end products (AGEs) in the trabecular meshwork in the lens, canal of Schlem, and optic nerve. This impairs mitochondrial water production, lowering Δψ and electrical resistance in retinal cells. This is how glaucoma, cataracts, and retinal damage all begin. It is not hard to see when you see it from 100,000 feet with the explanation in front of your eyes.
Outcome: Reduced photo-bioelectric in the anterior and posterio chamber signaling leads to RGC apoptosis and optic nerve degeneration, all hallmarks of glaucoma, independent of IOP in normal-tension glaucoma cases. They also lead to lens opacification.
Bioelectric Consequences: Dehydrated melanin in the orbit increases conductivity, potentially sending aberrant signals through CN V1 to the trigeminal nucleus and hypothalamus. This amplifies ROS/RNS and ultraweak biophoton transformation from the excessive bioelectric currents in the eye (per the work of Roeland Van Wijk and Fritz Popp references), further driving pathology in the central retinal pathways proximally and distally in neural structures and circulatory structures. This is how we get opthalmic aneurysms too.
Tinnitus Connection: The hypothalamus also influences the auditory system indirectly via autonomic pathways (e.g., trigeminal-autonomic interactions). Disrupted melanin in the orbit might contributes to tinnitus (as in Dr. Poland’s case) by altering photo-bioelectric signals that reach the brainstem and auditory cortex. Never forget spike protein is a mitochondrial toxin that mimics blue light and nnEMF damage.
SUMMARY
Cataracts’ cause a creeping blindness that disrupts light’s quantum role, damaging organs and perception via altered UPEs and epigenetics, not just vision. Light’s power depends on absolute darkness at night, and its blockage shifts human evolution toward disease unless addressed with wisdom and unconventional approaches. Modern centralized science’s irony, is that astrophysicists study stellar light without ever linking it to cellular evolution. This defect in methodology underscores the need to prioritize light-driven epigenetics over genomic dogma. Nothing in the literature is trustworthy because of this disconnect.
The real issue isn’t too much sunlight, it’s the wrong kind of light, at the wrong time, without the full-spectrum solar balance nature intended. You must get more sun, not less when your lenses are opacified. There are many pathways to cataract formation but excessive blue light and nnEMF exposure is by far the number one cause in the modern world.
The neural networks of the cornea and orbit converge in several brain regions. Still, the hypothalamus is the most likely central hub where a lack of melanin would have a significant impact in cataract formation, given its role in melanopsin signaling, autonomic regulation, and circadian control.
When you have a cataract, you have sustained a light injury to the hypothalamus. A cataract, therefore, should be thought of as a TBI, made chronically by low intensity nnEMF exposure of the eyes. This mimics an electric scar on the lens. The pathology all begins with vasopressin release from the light injury. Quantum Engineering #23 explains why this is the case. Please review it. Therefore, the decentralized clinicians should employ antagonists of the vassopressin signal. That would be sunlight with more red diurnally than expected because this light makes more DDW water from mitochondria.

This water is not as conductive to the exposed 30 million volt charge of the mtDNA exposed where melanin degradation has occurred. A melanin deficit in the cornea/orbit (or its functional dehydration due to nnEMF/ALAN) should be expected to disrupt hypothalamic functions, leading to autonomic and bioelectric imbalances that exacerbate conditions like cataracts, photophobia, or even tinnitus.
Many other conditions of the hypothalamus should be expected in these patients. Consideration for those with obesity (choroid/arcuate nucleus), Hashimotos (Ant pituitary), anorexia, ASD (thalamus), ADHD (habenular nucleus), altered perceptions of reality, poor first principle thinking, or gender dysphoria. The trigeminal sensory nucleus and thalamus are secondary regions where these effects might manifest as altered sensory processing disorders because their perception of reality is altered. This makes change hard for them to accept and perform.
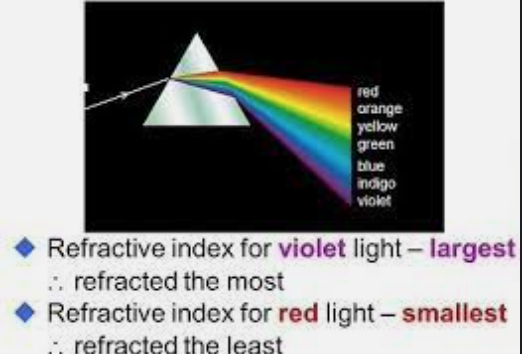
This blog should make you understand why I have rejected competition (Darwin’s “survival of the fittest”) for cooperation (Survival of the Wisest) and creativity aligns with epigenetic adaptability. Wisdom, gained through learning and error, decodes the “rainbow” in non-coding DNA and non-verbal communication in biology. Ignoring the centralized noise (genomic or verbal) so we can focus on the improbable (e.g., light’s evolutionary role) because when we do, this reveals nature’s secrets, as stars’ light evolves the entire universe. Cataract formation stops that process in you.
CITES
Are all buried in the substance of the blog. Review them all.