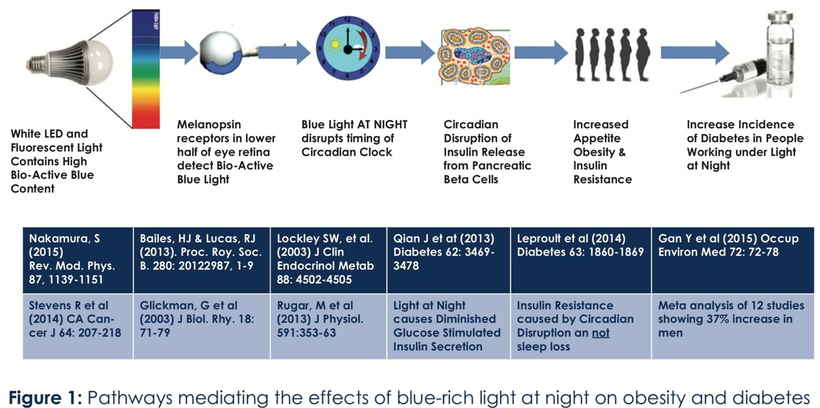
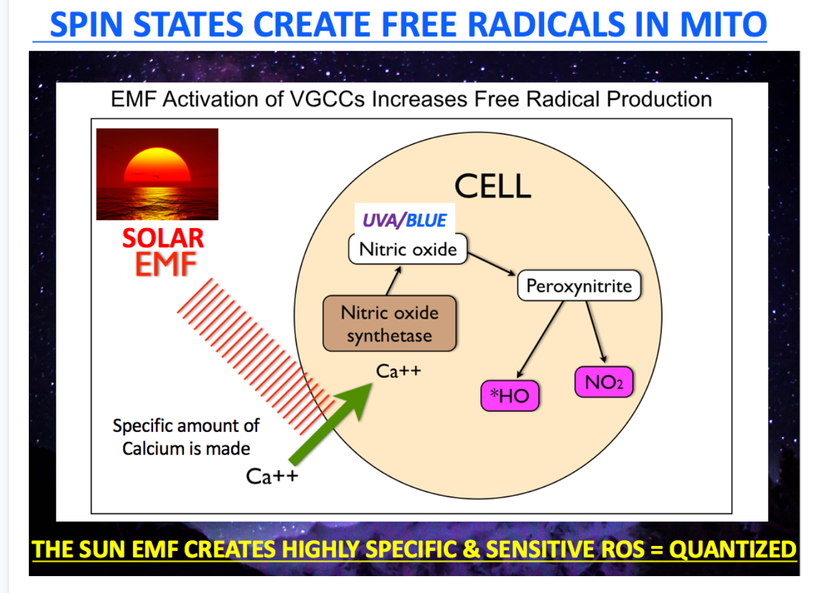
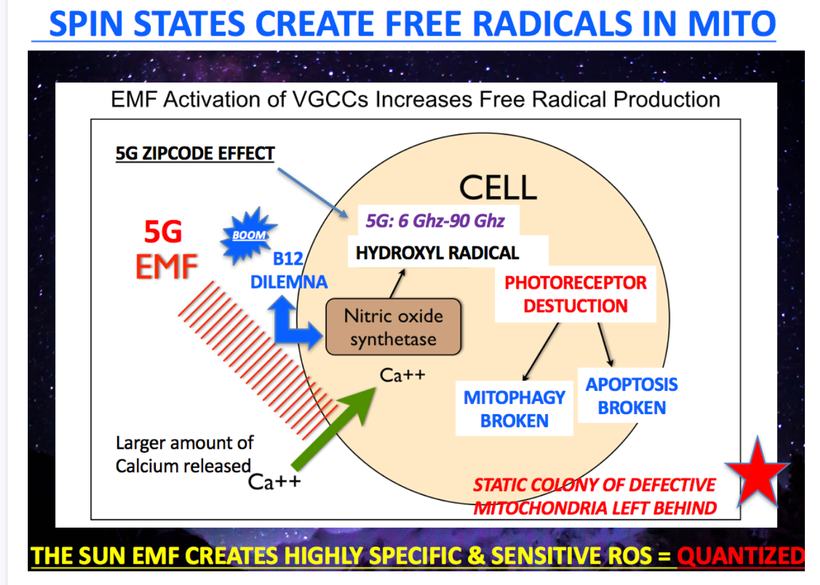
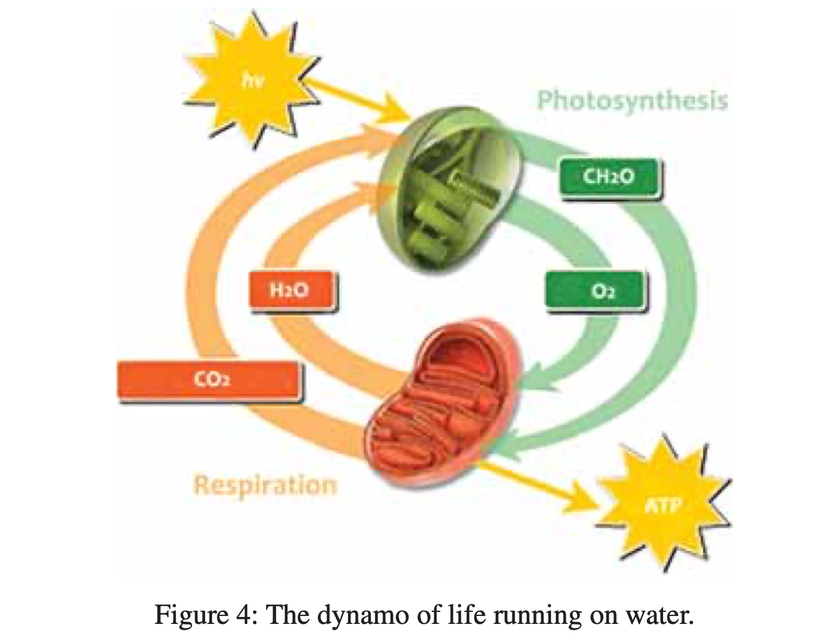
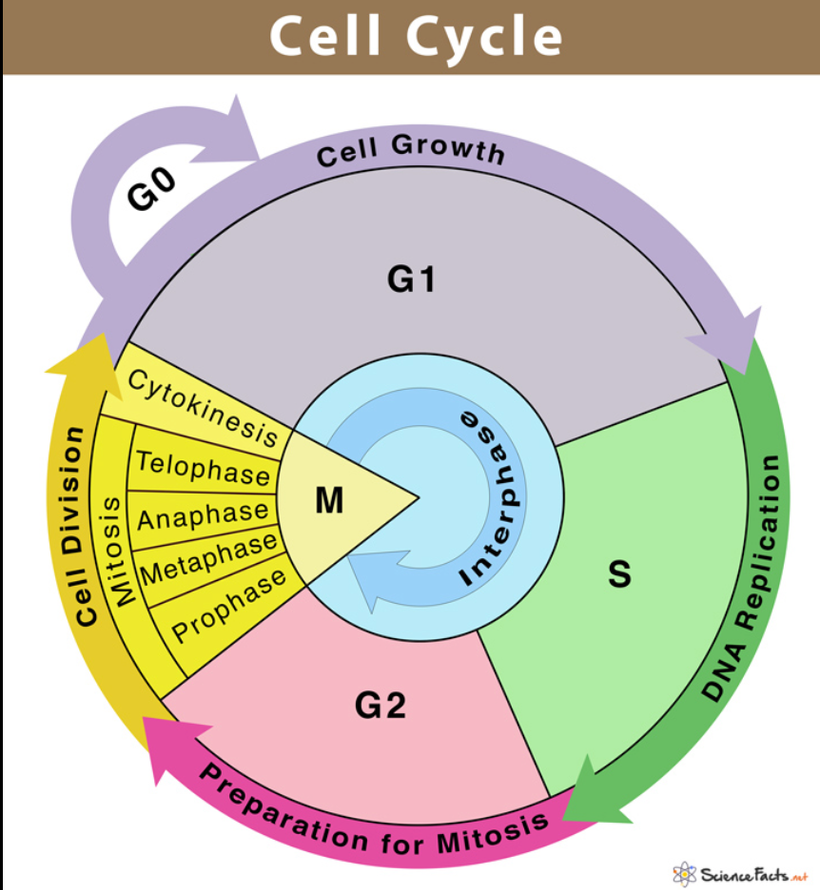
The future of maternal fetal medicine is understanding howthe governments MKULTRA programming has ruined the fundamental process.
Did you know the Leptin melanocortin pathway controls the entire physiology of the human placenta? This is why Leptin controls fecundity and fertility. The placenta mtDNA liberates more ultra-weak biophotons, which alter the hydrogen binding networks in the water in blood, and this liquid crystalline structure delivers its payload to the mother brain to begin translation of more POMC deposits in her brain. As water content increases in her circulatory system, so does melanin; this raises her alpha MSH level and increases melanin production in the brain. As a result of the relative rise of UV-powered water in her blood and increased melanin, her brain shrinks. This mimics the process I explained in my recent
Palestra Society talk in El Salvador last month. This phenomenon has allowed me to use cerebral brain volumes on my patients to predict transgenerational risks of the unborn child. This is how the future of maternal-fetal medicine will be practiced in decentralized medicine systems.
Nature always favors the living mavericks in her domain who challenge her rules. She will respond by fighting the maverick all the way, but she respects them and adapts to the environmental pressures the maverick applies. Ultimately, nothing else matters but the pressures placed, which cause her adaptations to occur in cells. They prove that she listens to them much more than Darwin’s ideas in centralized texts.
Morphogenesis develops during the first 5-6 months of pregnancy while the brain remains immature and devoid of development. Suddenly, the brain stops developing in six months, and the child gains more weight than at any other time of its in-utero life. How does this happen? The fetal mitochondria create light spectrum where U light is subtracted with IR light, and blue light is emitted. This fattens the child using melanopsin as its photoswitch. A LACK of endocabbinoidal function turns off the UV 380 nm mTOR photoswitch, and the IR 1280 nm photoswitch is also turned off.
This stunts brain development and fattens the child because there is no POMC translation; it results in a lack of melanin and endogenous endocannabinoid signaling. The green rectangle in the slide below shows you its dynamic effect. As leptin, adiponectin, and endocannabinoids drop, so does mTOR signaling. This means they must affect production of UV biophotons and they have to involved the mix of H+/D levels in the mitochondrial matrix.
Many forget that humans arrest neural development to be able to fit their offspring out of the vaginal canal safely. As neural development halts in humans, the child subcutaneous space needs to fattens up. That will facilitate the child’s passage through a tight canal in the mother, and the fat can be used to mature the brain when the child is born and is put in sunlight. 380 nm light not only matures the child’s brain post natally but also drives regenerative programming everywhere in humans. In fact, endogenous endocannabinoid signaling is critical in regulating decidual senescence and parturition timing.

380 nm light increase cognition in the developing brain. because it is associated with energy flow from matter to be turned on as the slide above shows. Synaptic proteins are upregulated also because of 380 nm because it increases nitric oxide (NO). People forget Nitric oxide (NO) works as a retrograde neurotransmitter in synapses. In brain it does different things than it does outside the CNS. NO allows the brain to increase blood flow to increase energy production and because of this it plays an important role in the fetal intracellular signaling in neurons to regulate the neuronal metabolic status to the dendritic spine growth. Remember as a free radical it controls the magnetochemistry of the fetal brain.
BPA increases deuterium absorption and reduces H+ flow in colony of mtDNA fetal brain. Deuterium in mtDNA is known to cause methylation defects, which causes endocrine dysfunction. Prenatal exposure to BPA at low levels can also affect gene expression in developing brain by turning off growth leading to cognitive decline. Fluoride does the same by stealing electrons that have these light frequencies in the UV range and this limits semiconduction and growth of the immature brain as it develops.
HOW DOES MOM’S PLACENTA FATTEN HER BABY PROPERLY?
Did you know the germinal matrix center in the human brain is found in the area of the brain called the thalamus? The thalmus is where all 5 of our sense target. I told you in the Quantum engineering series that all life has a frequency modulated antenna system built into the thalamus to tell time. Specifically it relates to telling seasonal time. It operates by varying the amount of deuterium in this part of the brain to alter its stiffness. Why?
This systemis built to vary its”flexibility” when it is encumbered by too much mass in the antennae of the system. This is where the Kruse for Dummies lecture comes to into play to explain to you how ATAVISM happens in utero to stop brain growth. It is instructive of why obesity in the adult form is a predictor for nnEMF risk. Humans are designed to fatten when they add deuterium to their thalamus to affect of the organism accounts for timing.
What bends space/time in the universe? The masses associated with the matter in the universe do. As result, physics teaches us that magnetic fields tend to flatten and stiffen the fabric of space-time when masses are added to them and this alters our perception of timing. So adding deuterium to the mitochondria of the developing thalamus stops neurogenesis in utero. The added deuterium alters our perception of the seasons, and this fattens the child in the third trimester. The process also halts melanin by turning off UV biophoton production in the placenta. This limits the neuroplasticity in the neural crest cells in different parts of the CNS of the fetus. If you remember from Becker’s work he said he found that the semiconductive pathway in neurons that controlled regeneration seems to come from just below the myelin level. This area is where POMC is normally found in humans and when UV biophotons are present would cause POMC translation to make melanin to charge separate water to make a ton of electrons that would signal a semiconductive circuit. When you turn off UV biophotons creation from mtDNA in the placenta and in the germinal matrix center you affectively stop brain growth.
Anything that has more mass than the smallest atom used inside of our molecular clocks changes how we experience time. This is AXIOMATIC. Mass bends space/time! As a result, we should expect the Fo head of the ATP to stop spinning as much. In fact, ATP production should slow down. How does this happen in humans. The mother’s placenta make a huge amount of NO to inhibit the fetal ATPase. This means that the magnetic fields created inside the childs developing neurons is effectively arrested. Anytime humans are using the TCA cycle, they are creating the largest magnetic fields in mtDNA.
Now ask yourself what magnetic fields do to space-time based on this picture below. It orders atoms around it to allow them to direct energy flow to create life. This process slows light down in tissues and causes time perception to change. The collapse of the magnetic field by the mother’s placenta halts this process. Centralized OB/GYN has never understood why the human placenta normally enhances physiological ROS production during pregnancy. Now you know why. It is not a bug in the design, it is a design feature to stop the childs brain growth.

What does this concept imply? Time slows as one approaches the speed of light. At the speed of light, time ceases to change because it contains all change.
A mother that does not get enough sun on her abdomen during pregnancy runs the risk of developing preeclampsia (PE). PE pathophysiology result from abnormal placentation due to a defective trophoblastic invasion and an impaired remodeling of uterine spiral arteries, leading to a poor adaptation of utero-placental circulation. Leptin and melanin control this process. This can alter how the brain develops in her child. This is really how autism begins in humans. It is also how many childhood cancers begin and I believe it is where Angelman’s Sundrome really comes from. Altered circadian biology of the mother’s placenta combined with a altered germline in eggs and sperm.

Note on the slide above that matenral levels of Vitamin D links to NO production of the placenta. This is why maternal Vitamin D levels are a huge predictor of problems in decentralized medicine. Mother’s have to use their stored light to turn off the magnetic fields in their child. People have totally forgotten the basics. Nitric Oxide turns off CCO and ATP production as the slide above shows. NO enhances blood flow through the placenta but it turns off energy flow to control the child’s brain growth. Fetal RBC contain mitochondria to make them less able to tell time so the child does not get involved with the mother’s placental signal to light. So having a mitochondria in your RBCs means you are less of a sensor to the SCN in the retinohypothalmic tract. This means the placenta of man controls the timing mechanisms in the baby when it makes sense to turn off brain development.
It implies that the your skin and eye take over this process when you leave the uterus as well.
WHERE DOES THE METHYLATION PROBLEMS COME FROM?
Tetrahydrobiopterin is a cofactor of the three aromatic amino acid hydroxylase enzymes that I have showed you hundred fo times in the slide below. THB is also used in the degradation of amino acid phenylalanine and in the biosynthesis of the neurotransmitters serotonin (5-hydroxytryptamine, 5-HT), melatonin, dopamine, norepinephrine (noradrenaline), epinephrine (adrenaline), and is a cofactor for the production of nitric oxide (NO) by the nitric oxide synthases.
I just told you the placenta controls NO production, didn’t I. Have you forgotten how NO controls methylation production? An altered antioxidant capacity from the mother’s circadian defects alteres her NO bioavailability in her placenta. Without NO can you collapse the ATPAse? If you cannot do this, can you control morphogenesis or organogenesis in the fetus? NOPE.
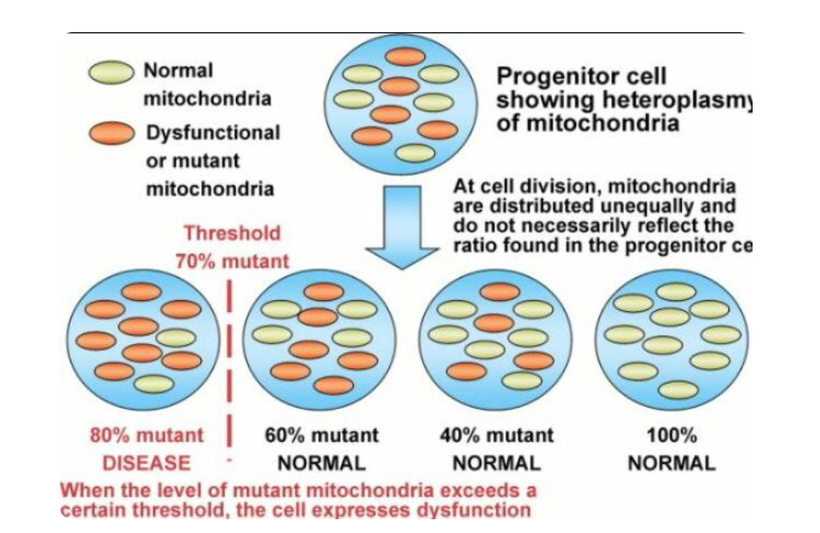
Altered NO levels leads to methylation problems in the nucleus. Centralized science has forgotten the basics. Why is this true? Altered methyaltion results in part from the reaction of NO with the radical anion superoxide (O2•−), which produces peroxynitrite ONOO-,. This is a powerful pro-oxidant and inflammatory agent. Another mechanism is the progressive inhibition of the placental endothelial nitric oxide synthase (eNOS) by oxidative stress. DIdn’t I tell you over a decade ago in the Holy Trinity blog that eNOS is a key circadian switch in the SCN of all mammals? I did. What happens when you affect it? An altered eNOS results uncoupling viaseveral events such as a depletion of the eNOS substrate L-arginine due to increased arginase activity, an oxidation of the eNOS cofactor tetrahydrobiopterin (BH4). It also implies an alteration in eNOS post-translational modifications (for instance by S-glutathionylation). This is how childhood disease manifest in utero. This is how a child is born with a large amount of heteroplasmy when the system signals are not well controlled by sunlight.
More heteroplasmy = higher disease burden in a fetus even before it has taken a breath.
How big a miss is this decentralized science for every centralized OB/GYN?
Did you know that tetrahydrobiopterin is a cofactor for tryptophan hydroxylase (TPH) for the conversion of L-tryptophan (TRP) to 5-hydroxytryptophan (5-HTP)? Tyrosine hydroxylase (TH) catalyses the conversion of L-tyrosine to L-DOPA (DOPA), which is the precursor for dopamine. You do remember that dopamine and melantonin control ALL RENOVATIONS OF PHOTORECEPTORS RIGHT? How many times have you seen this slide as proof I have been telling you this?

Now what are all those photoreceptors I keep mentioning? Remember how many times you have seen this slide in blogs too. The same message has been being pound into your blue light toxic brains for decades.

The control of THB and NO by the placenta turns out to be critical in melanin production as the slide below shows. This is the real reason why most centralized OB/GYNs have to give women prenatal vitamins with folic acid. WHY?

BH4 can be oxidized by one or two electron reactions, to generate BH4 or BH3 radical and BH2, respectively. Research shows that ascorbic acid (also known as vitamin C) can reduce BH3 radical into BH4 preventing the BH3 radical from reacting with other free radicals like superoxide and peroxynitrite specifically. Without this recycling process, uncoupling of the endothelial nitric oxide synthase (eNOS) enzyme and reduced bioavailability of the vasodilatornitric oxide occur, creating a form of endothelial dysfunction. This is the only thing centralized OB/GYNs got right. They realized that a lack of folic acid was linked to caudal regression syndromes in fetal medicine. The problem is they still have no idea it links to just about every other disease children get from solar deficit mothers. Ascorbic acid is oxidized to dehydroascorbic acid during this process, although it can be recycled back to ascorbic acid to be reused. Look at the slide above. You’ll see how the placenta turns off the recycling of Vitamin C and now you’ll see why. Glutamate release without Vitamin C turns off neurulation in humans.
Folic acid and its metabolites seem to be particularly important in the recycling of BH4 and NOS coupling. This is why this series has a blog on this. When mothers’ are solar deficient they will need folic acid to prevent many diseases in maternal fetal medicine.
I know you’ve seen this slide below a lot too., but I need to you to connect the lessons I am stacking for you now because centralized medicine is being sculpted by BigHarma to make sure you do not get this lesson via how the curriculums in med school have been hijacked by data that came from MKUTLRA. Blocking this pathway on the top line of the slide below stops brain growth in the fetus!!! It also stops T3 production which is needed for neuron growth and sprouting in humans.

What else is caused by this placental light dysfunction story I am weaving for you?
Phenylalanine hydroxylase (PAH) catalyses the conversion of L-phenylalanine(PHE) to L-tyrosine (TYR). Therefore, a deficiency in tetrahydrobiopterin can cause a toxic buildup of L-phenylalanine, which manifests as the severe neurological issues seen in phenylketonuria. So PKU is a risk factor for women who are solar deficient. I bet your doctor never told you that!
HOW WE CREATE DIABETICS?
The role of BH4 in this enzymatic process is so critical in arresting fetal brain growth that most have forgotten that a deficiency of BH4 in the placenta means – WE SHOULD expect a reduction of nitric oxide production. This is the photoswitch that controls this process. Without BH4 you cannot make NO at all. Recall that Nitric oxide synthase (NOS) catalyses the conversion of a guanidino nitrogen of L-arginine (L-Arg) to nitric oxide (NO).
Why does maternal fetal NO causes gestational diabetes? It should be obvious now. Moreover, it should be obvious why gestational diabetes means the mother and baby are going to be future diabetics. Why do I say this? 30 years of maternal fetal medicine research has pointed to a deficiency of BH4 – and thus, of nitric oxide – as being a core cause of the neurovascular dysfunction that is the hallmark of circulation-related diseases such as diabetes. Anyone who is solar deficient and gets more ALAN is guarranteed to get diabetes because of a chronic lack of NO production. Moreover, why do diabetics get more cancer? NO controls the stem cell depot biology. And a lack of BH4 also causes DNA methyaltion defects which make oncogenesis more likely. EVERYTHING FITS DECENTRALIZED medicine theory here, doesn’t it?

IF YOU BLOCK THE SUN YOU WILL GET CHRONIC DISEASES IN EPIDEMIC FASHION.
I hope youre beginning to understand how atavistic effects predict the future of mankind now as they built a tech world with MKULTA light.
Your mother’s placenta uses light message from her mtDNA to create a defect in the antenna buried in your developing thalamus where all human neurogensis occurs. This optical lattice clock switch in her placental ruins the fidelity of the system in the fetal brain by design so it can no longer tell where the Earth and sun are in relation to one another. It destroys the ultradian ryhythms in the child by design.
FOOD mom eats has zero to do with this mechanism. Her SCN sends this signal to her mtDNA in her placenta. Her SCN is an optical lattice clock; it is not a food clock. . It is a pretty remarkable gadget Nature built in our heads and our placenta.
It appears the simple addition of a neutron to H+ to the antenna system in the fetal thalmus is enought to screw up the connection of the thalmus to the heartbeat of the Earth. Without a proper connection to Earth, the fidelity of the music coming from the system fails. What does this FM station listen to? The Schumann resonace of the Earth. It is 7.83 Hz. That is the human alpha wave which is created in the thalamus. That is what causes all chronic disease in humans. It turns out adding neutrons to H+ is enough to do something to the fabric of space-time in the fetal brain that deforms the magnetic fields in the child’s ATPase to allow a child to emerge from a small pelvic outlet. This shows you how the smallest things make the biggest deal in you. Wild decentralized FACTS, that have been blocked in centralized science, I tell you. Sadly this science was shared with the corporations that were involved with the Industrial military complex in the 1970’s and 1980s.
THE PHYSICS WAS WORKED OUT FOR ME IN THE BASEMENT OF CHARITY HOSPITAL IN 1989.

The spacetime curvature for a charged static spherical body is given by the Reissner–Nordström metric. I’d suggest you carefully look at the next slide many many times. It shows you what Einstein’s special relativity really shows. This was critical for the development of technology to operate wirelessly using satellites to control them above Earth. This is why the GPA in you iphone actually works like your SCN does.
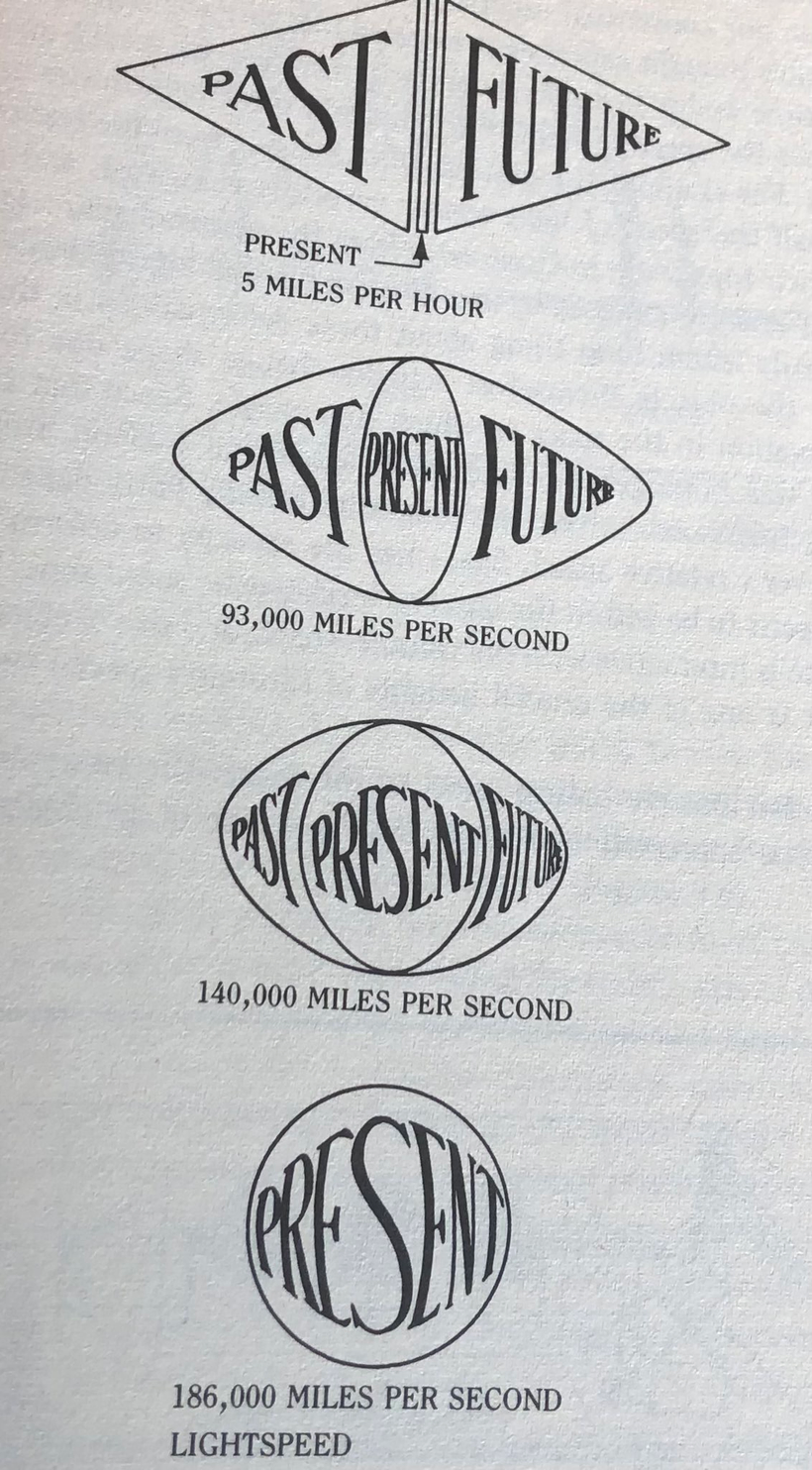
The picture above shows you time slows as one approachs the speed of light. The present moment your living right now reading this expands from a narrow sliver until it encompasses both the past and the future. This means that the idea of past and future is an optical illusion caused by the slowing of light by matter. It also means as light slows you get the idea there is a cause and effect on Earth but there is no real relationship. This is why BigHarma continues to use the idea that RCT is the gold standard for centralized science. The physics of special relativity tell you this is a bullshit story from the jump.
When you use these mathematics, you can feed in the value of whatever charge you want and calculate the time dilation as a function of distance from the charged body. If you do this you’ll discover something rather odd in Einstein’s field equations, namely, the charge reverses the effect of the mass in them.
This is why REDOX POTENTIAL is the key to chronic disease reversal and nothing else matters.
READ THAT LAST SENTENCE AGAIN.
This message was given to the Industrial military complex Big Pharma partners. They were told to add atoms to their drugs to limit the effect of charge. This helps the military use light to control people by using screen technology. It stole their dopamine reward tracts. I mentioned this pathway above. REVIEW IT.
That is what the target of MKULTRA was focused on after all the experiments where completed from 1953-1964. Those experiments continued until the early 1980’s by the CIA In Reagan’s admiistration. It was given to researchers who linked colleges via the ARPANET. This data was them shared by the Industrial military Microsoft’s Bill Gates and Apple’s Steve Jobs by DARPA in the 1980s. This was done by DARPA but pre- GOOGLE. . All that data was sitting in the basement of Charity Hospital where I did my residency. It all came from Tulane neurology and neurosugery experiments done for MKUTRA and by Sidney Gotlieb. I read them all before it was all destroyed by Huricane Katrina. The basement flooded and almost 160 years of medicine was lost in New Orleans.
How do cells increase their charge?
LIGHT DOES IT. Food has no effect on this.
Read it again.

LOOK MORE CAREFULLY AT THE SLIDE ABOVE AGAIN. It is why I posted it again. Do you remember how many times I have told you in podcasts that redox potential is formed by adding net negative charges in a cell? Melanin is the key way all mammals do it. Human biology relies heavily on this. Do you see now by examining the slide above closely how that charge in light frequencies can offset the bad isotopic variation in some of your tissues to make your clocks work better in the circuit that tells the brain what season it is? This is another universal law not subject to an RCT. BigHarma knows what your doctors do not. And they are not going to tell them either. It is encumbent upon you in getting this lesson and why it is being blocked from centralized MDs. It should explain to you why the Universities in ARPA founded HMOs and changes the way healthcare was delivered in the 1980s to put an Oracle between you and your doctor. That Oracle was a healthcare industrial complex player. The system was engineered to put a hospital CEO and an insurance executive decision tree between you and your doctor before ANYtreatment plan can be APPROVED by the governments agents in this plan. HMOs and the insurance industry were designed to limit the time a doctor can spend with you so you only have time to get a presciption that comes from BigHarma that changes the charge in you so you are more likely to follow the rules built by government. I will remind you that Medicare began post MKULTRA in 1968 under LBJ post JFKs assassination.
HOW WAS MKULTRA, LBJ, THE CIA, JFK, AND ISRAEL ALL LINKED TO THIS STORY?
I laid most of it out in the recent Danny Jones podcast but i saved the critical details part for my members so you can understand how it links to the creation of all modern disease epidemics.
THE MOST EXPLOSIVE SUMMARY
On July 30, 1965, President Lyndon B. Johnson signed into law legislation that established the Medicare and Medicaid programs. This created CMS for the industrial healthcare complex. It established Medicare, a health insurance program for the elderly who could and would be taken out, and Medicaid, a health insurance program for people with limited income to make sure they remained poor and sick to be controlled by the government for decades to come. LBJ signed this law in Indepence, Missouri. Why? This is where Manchurian Candidate Harry S. Truman was from.
This completed the medical tyranny portion of the New Deal Era that began under FDR and was linked to the the Manhattan Project story of how DARPA was created by General Groves. How long ago did the Industrial military complex decide to use medical tyranny as the attack vector of the US Constiution? 1911.
This was when the Flexnor report was launched to create BigHarma from Rockefeller’s break up of Standard Oil by Teddy Roosevelt. I had a lot to say about this in the Danny Jones podcast, but I will have a lot more to say about it in future podcasts once this information in that podcast is married in this summary and understood by the public.
Today, you are being highly preconditioned for compliance by the real findings that came from MKULTRA.
At the same time that CMS was created the ideas to taper the Ponzi scheme of money laundering from the public, the USA Executive Branch and the CIA helped pay off the MOSSAD for the JFK assassination by giving Isreal close to 300 pounds of uranium it needed to become a nuclear power. JFK, prior to his death, was going to send in the Atomic and nuclear regulators of the USA in to make sure Isreal never got an atomic weapon. This infuriated Israeli prime minister David Ben Gurion.

CAREFULLY EXAMINE THE PICTURE ABOVE AND NOTE THE DATE IT WAS TAKEN.
How did this story link JFK to Israel and the MOSSAD?
It began with an intelligence failure of the CIA and John Kerry’s father.
Documents published in Wilson Center from the National Archives now have shed light on a particularly notable intelligence failure: how Washington missed warning signs that the Israelis had a nuclear project underway, but also how the U.S. belatedly realized what the Israelis were doing, and how Eisenhower and his senior advisers reacted to this discovery. Among the new documents are:
- The June 1959 Israel-Norway secret agreement providing for the sale of Norwegian heavy water to Israel (through the United Kingdom), transmitted by Oslo Embassy political officer Richard Kerry (father of former Secretary of State John Kerry).
- Reports about information from a then-covert source — University of Michigan nuclear engineering professor Henry Gomberg — who learned that the Israelis had a secret nuclear reactor project that involved experiments with plutonium.
- A telegram from the U.S. embassy in Tel Aviv reporting on Finance Ministry official Addy Cohen’s statement that “we’ve been misbehaving,” and one by an unidentified official close to Prime Minister David Ben-Gurion that the secrecy surrounding Dimona was unjustifiable and that it was “a stupid mistake on the part of Israel.”
- Reports by U.S. Ambassador Ogden Reid on conversations with Ben-Gurion.
- A State Department message to the embassy in Tel Aviv conveying irritation that the responses of the Israeli government showed a “lack of candor.”
- Messages about a role for the International Atomic Energy Agency in inspecting and safeguarding Dimona. When Eisenhower left office Kennedy assumed this mess but he knew Ben Gurion for over a decade as the picture above shows. Now President Kennedy pressured the government of Prime Minister David Ben-Gurion to prevent a military nuclear program, particularly after stage-managed tours of the Dimona facility for U.S. government scientists in 1961 and 1962 raised suspicions within U.S. intelligence that Israel might be concealing its underlying nuclear aims. This went on during the Cuban Missle crisis and Kennedy was worried about nuclear proliferation post WW2. Kennedy’s long-run objective, documents now show, was to broaden and institutionalize inspections of Dimona by the International Atomic Energy Agency.
Of all U.S. leaders in the nuclear age, John F. Kennedy was the nonproliferation president. Nuclear proliferation was his “private nightmare,” as Glenn Seaborg, his Atomic Energy Commission chairman, noted many times in interviews. Seaborg discovered Plutonium for the Industrial military complex leaders.
On 30 May 1961, Kennedy met Ben-Gurion in Manhattan to discuss the bilateral relationship and Middle East issues. However, a central (and indeed the first) issue in their meeting was the Israeli nuclear program, about which President Kennedy was most concerned. According to a draft record of their discussion in the National Archives, which has never been cited, but has now been released for the first time by the Wilson Center, Ben-Gurion spoke “rapidly and in a low voice” and “some words were missed.” He emphasized the peaceful, economic development-oriented nature of the Israeli nuclear project.
Nevertheless the note taker, Assistant Secretary of State Philips Talbot, believed that he heard Ben-Gurion mention a “pilot” plant to process plutonium for “atomic power” and also say that “there is no intention to develop weapons capacity now.” This information stunned JFK. Ben-Gurion tacitly acknowledged that the Dimona reactor had a military potential. The final U.S. version of the memcon retained the sentence about plutonium but did not include the language about a “pilot” plant and “weapons capacity.” Anyone who understands basic nuclear physics (see my Patreon Thorium blog on BTC) knows the only reason to use plutonium as a fuel is because you can make military grade uranium for nuclear weapons.
The differences between the two versions suggest the difficulty of preparing accurate records of meetings. But whatever Ben-Gurion actually said, President Kennedy was never wholly satisfied with the insistence that Dimona was strictly a peaceful project. This really was the main precursor to his death in my estimation. Neither were U.S. intelligence professionals. A recently declassified National Intelligence Estimate on Israel prepared several months after the meeting concluded that “Israel may have decided to undertake a nuclear weapons program. That CIA information was leaked to the mob in 1963 after the Bay of Pigs invasion went south. This information was critical to Santo Trafacante because it allowed him ot gain leverage over Castro and the attempts of the Industrial military complex to kill him versus JFK. Trafacante ensured it was JFK that would pay the ultimate price. He viewed his appointment of RFK Sr as Attorney General as the ultimate act of betrayal to the mob bosses in Chicago, New York, Florida, and New Orleans. In fact, Carlos Marcello was on trial as JFK was shot in Dallas on November 22, 1963 because of an RFK Sr prosecution that Joe Kennedy swore would never happen if the mob swung the swing state of Illinois to JFK in 1960. These details matter big time to understand how things really unfolded.
ISRAEL had massive amounts of agents in the USA in the Jewish mob before Israel was even made a country in 1948. Prior to being a country they had a massive intelligence capability world wide that linked banking, science, and gangsters who were all Jewish.
This was the main reason the Jewish gangsters were eventually made MOSSAD agents after 1948. They fed information to Israel that the Chicago and NY families got on Joe Kennedy and his sons. As a result, Meyer Lansky, Jack Ruby Rubenstein, Carlos Marcello, and Santo Trafacante became involved with the assassination of JFK , along with Nixon, the Bush and Hunt Oil families and Clint Murchison linked to Groves Industrial military complex. Texas was also a swing state and the Oilmen were pissed that Kennedy was catering to Blacks in civil rights and was soft on the Communists in Russia and Cuba.

Santo Trafacante was among the most powerful Mafia bosses in the United States when JFK was a Senator in the 1950s. He headed the Trafficante crime family from 1954 to 1987 and controlled organized criminal operations in Florida and Cuba. He was also a MOSSAD asset for reasons I laid out in the Danny Jones Podcast. He was a double agent for the CIA and MOSSAD in the 1960s due to drug laundering to replace what the Chicago and Florida families lost in the casinos due to the Cuban Revolution of 1959.
Castro enlisted Santo to pay back the CIA for trying to kill him 9 times by joining forces with Traficante to turn Americans into drug zombies. Trafacante was the key link the Italian mob to the Jewish in the USA. This is how the MOSSAD got the information that JFK life was at risk. He also was in charge of monitoring the behavior of Judyth Vary Baker under Dr. Alton Ochsner care on Magazine St in New Orleans. He also linked the Chicago mob the NYC mob of Bonanno crime family. This family is known as the American mafia. They are constructed of 5 families all based in New York. These Families had connections with FDR Jr son as a Senator from NY.
Carlos Marcello was a gangster who was based in New Orleans but tightly linked with Sam Giancana of Chicago. Both worked with Trafacante in Florida and Chicago. He knew Lee Harvey Oswald since Oswald grew up in New Orleans. In 2022 more government documents were released which showed us how US law enforcement was linked to the Italian mob. At least nine files released refer to Chicago circa 1963 and several individuals linked to the Outfit.

Most notably among them are former Chicago police officer and Cook County Deputy Sheriff Richard Cain (see above). Cain had deep connections to Chicago organized crime according to investigators, especially ruthless mob boss Sam “Momo” Giancana. Joe Kennedy, father or JFK, asked for Sam’s help in getting Kennedy elected in 1960 through his connections with labor unions linked to Jimmy Hoffa. Illinois was a He was promised under JFK presidency there would be a laissez faire action plan towards organized cirme. This is why Sam helped Kennedy’s father. It is clear today that JFK or RFK was not given this information. It is also clear that RFK Jr knows this information today.
In 1965, the MOSSAD was paid off when Isreal got uranium from LBJ and CIA as payment for their assistance. Did Israel steal bomb-grade uranium from the United States or were they given it by the USA?
After a 1965 inventory, NUMEC was found to be missing about 100 kilograms of bomb-grade uranium, even after accounting for all processing losses. The close personal and commercial ties to Israel of the plant owners and operators raised suspicions that remained unresolved. The affair of the missing bomb-grade uranium was revived in 1976. The newly formed NRC in 1976 UNDER fellow Warren Commission member Gerald Ford who replaced Nixon in 1974.
The NRC was in the process of writing licensing regulations for commercial fuel firms—of which NUMEC was one—and had heard rumors of possible theft in the 1960s from NUMEC’s Apollo facility. NUMEC = Nuclear Materials and Equipment Corporation. NUMEC is based in Pennsylvania. There is a reason this plant was targeted by LBJ, CIA, and Israel. The partners in the JFK crime syndicate were in control of this location. Arlen Specter the lawyer who came up with the one bullet theory in the Warren Commission was given the Senator postion by the Industrial military complex by using election stealing capabilities in the 1960s. The same ones were used to give the election of JFK over Nixon in Illinois and Texas in the 1960 election.
The NRC asked for a CIA briefing. This was by design because President Ford knew the CIA was involved in the theft to pay Israel off. Duckett startled the NRC group with CIA’s conclusion that the missing uranium was in Israeli bombs. The NRC chairman informed the White House, and President Ford took an iimmediate nterest in the case because Duckett was not supposed to tell the truth. He was not involved in the JFK assassination.
Ford’s Attorney General, Edward Levi, discovered that the Atomic Energy Commission (AEC), the NRC predecessor nuclear licensing agency, had previously convinced the FBI not to open a criminal investigation into the material’s disappearance. Why would he do this? Levi was the first Jewish Attorney General of the United States. He was also a MOSSAD asset.
Levi was a Yale Graduate with ties to Israel. In fact, he was a Sterling Fellow at Yale and that dates his affliation with Israel all the way back to 1864 and B’nai Birth. B’nai Birth is the previous group now called AIPAC. Before AIPAC they were the ADL. What is the history of B’nai Birth? This group was loyal to the Royal Family before the Civil War in the USA. As a Yale graduate he was linked to the Bush oil family who was an early partner of the Industrial military complex General Groves put together from the ONI and OSS before the CIA was formed. Preston Bush, the father of George H Bush, was a banker who worked Brown Harriman in NYC who worked with the families who controlled money in the US Federal Reserve. This was established in 1911.
During Levi’s term as Attorney General, he issued a set of guidelines in 1976 to limit the activities of the FBI. This was done in repsonse to Nixon being forced to resign over wiretapping during Watergate. The CIA was furious that the FBI turned on Nixon and this was one way the DOJ in the executive branch was used via lawfare. These Levi guidelines required the FBI to show evidence of a crime before using secret police techniques like wiretaps or entering someone’s home without warning.
How connected to the criminal cabal was Levi? His guidelines were QUICKLY replaced by new ones issued in 1983 by Ronald Reagan’s Attorney General, William French Smith. Levi urged President Ford to appoint Robert Bork, who was his former student and Solicitor General to the SCOTUS. The real reason Bork was not elevated to the SCOTUS is because many democrats knew that Levi and Bork were linked to the conspiracy that assassinated JFK. That did not stop Levi. He told Ford if Bork is blocked he should nominate fellow Chicagoan John Paul Stevens to the United States Supreme Court, and Ford followed his advice. Did you know that Levi later testified in support of Bork at his confirmation hearing for the SCOTUS? Did you know who else was linked to Levi and Israel?
Serving under Levi at various times in his career, in various high staff positions, were such people as Rudolph Giuliani, Robert Bork, Antonin Scalia, Rex E. Lee, and Arthur Raymond Randolph. That is how close the ties with Israel and the MOSSAD are to the USA. It is also why I told Danny Jones why Donald Trump made the JFK achives public. As president, he found how his friends in banking, Wall Street, and real estate were to the JFK assassination attempt. They are the people behind the warning shots being made right now to DJT. I believe if they wanted him dead, he’d already be dead. I think they think they can control Trump if they make it clear to him during this election cycle that he needs to follow the playbook Levi built out in the 1970s. Joinign forces with RFK Jr is why they went after him at Butler in Penssylvania. It should not surpise anyone of you now why it happened in Pennsylvania. The industrial military control over this state and it politicians is close to air tight.
HOW TIGHT IS PENNSYLVANIA in this story?
I told you that NUMEC is in Pennsylavania. Did you know that Zalman Shapiro the owner of NUMEC attorney was Arlen Specter, the Senator for Pennsynvania? Amazing coincidence or was the something more important to this story the USA government wanted buried forever?
So the chain of evidence went from Ford to Bush to Carter to the CIA or did it?
Carter instructed his national security advisor, Zbigniew Brzezinski, the father of modern day broadcaster Mike on MSNBC, to deal with the NUMEC matter in the context of the impending public release of MUF data. Brzezinski’s staffers John Marcum and Jessica Tuchman posed questions to the CIA about the NUMEC affair. This is when key data magically went missing to President Carter.
WHAT WENT MISSING?
Carter was never told about the links of Israel to NUMEC.
Brzezinski shared the information with 2 staffers but not Carter. The two staffers told Brzezinski that environmental samples taken by the CIA in Israel in 1968 contained highly enriched uranium, whose enrichment level was so high it pointed to the Portsmouth, Ohio, uranium enrichment plant as the source. Portsmouth was where NUMEC obtained uranium stock for its naval fuel products. Why he never told Carter is likely tied to his ideology as Polish and hating Russia and this marries to General Groves ideology linked to Stalin.

During the Apollo affari of 1965 the president of NUMEC, Zalman Shapiro, had frequent contacts with Israeli officials, including a science attaché “thought to be an intelligence officer,” and received unexplained VIP treatment in Israel. None of this information went from Brzezinski to Carter.
In fact, Shapiro was by then known to have had contacts with Israel’s head of military intelligence and the head of its nuclear weapons program. He later acknowledged knowing Binyamin Blumberg, head of Israel’s “bureau of scientific liaison,” which engaged in high-risk intelligence capers. On one strange occasion in 1968, Shapiro hosted an Israeli intelligence foursome at the Apollo plant. One was the Mossad agent who headed the team that spirited former Nazi leader Adolf Eichmann out of Argentina and who later ran Jonathan Pollard’s spying on the United States for Israel.
For those of you who do not know, the Mossad is the Israeli agency that handles foreign intelligence collection and covert action. It is Israel version of the CIA. Another was that agent’s deputy in the Eichmann kidnap, who went on to become head of Shin Bet, Israel’s internal security service. A third was Mossad’s director of technical services. The last was Israel’s science attaché, who had held a senior position in Israel’s nuclear weapons program. More amazing coincidences in this story do not you think?
How did I know all this history and link it as a young man before I became a doctor in New Orleans? The man who i have spoken about in many podcasts who taught me everything I know about fiat banking and fractional reserve banking was named Theodore Sterling of the Sterling National Bank on 57th Street and Madison Ave as I grew up in NYC. He told me this history when he told me how banking really operates in the USA. Read the hyperlink on him you just passed before going further.
What happens when Ford leaves office? How did the story remains toghtley controlled by the industrial military complex?
Everyone involved was linked to the Warren Commission.
After the 1976 election, the Ford White House alerted the incoming Carter administration to the NUMEC affair. In December 1976, according to a July 1977 National Security Council memorandum, then-CIA Director George H.W. Bush briefed President-elect Carter on the case. Congress had been pressing for public disclosure of records of large unexplained losses of bomb-grade material (in government parlance, “material unaccounted for,”or MUF) from the government’s nuclear weapons complex. The Carter White House feared the story was sure to hit the headlines if there were any suggestion of Israeli theft from the NUMEC facility.
And any disclosures about Israel’s bomb program would of course have threatened the Carter administration’s Middle East policies between Israel and Egypt. If you really want to know the truth, I have always believed Israel agreed to this peace deal reluctantly to bury the Apollo affair once and for all. But that affair is the gift that keeps giving. It’s secrecy is linked to the real story of how the MOSSAD was involved in 9/11 Tower knockdown fiasco during the George W. Bush presidency. See how the story all begins to make sense when you really see it laid out as it happened. Today’s history books will never let you see how it is all linked.
MOST OF YOU HAVE NO IDEA HOW NEFARIOUS OR HOW OLD THIS PLAN IS IN THE HISTORY OF THE USA.

CITES
1. Danny Jones podcast
2. https://thebulletin.org/2014/04/did-israel-steal-bomb-grade-uranium-from-the-united-states/
3. https://abc7chicago.com/jfk-files-documents-national-archives-chicago-mob/12576586/