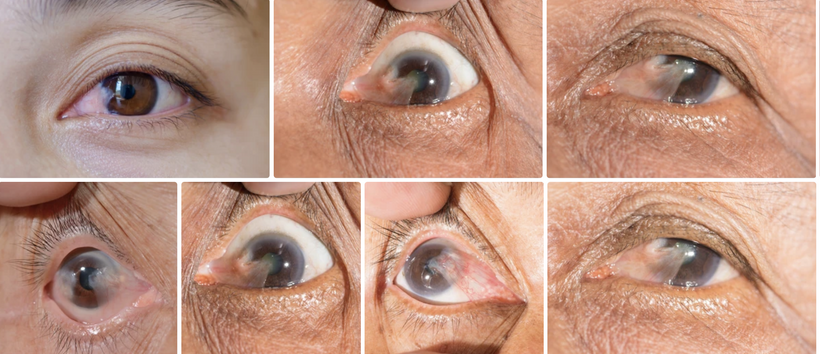
I propose that anemia of chronic disease (ACD) is a blue light toxicity problem, related to the Great Oxygen Holocaust due to our technocracy. It stems from melanopsin damage in the eye and skin and affects heme protein production in the bone marrow. This is an extension of my photo-bioelectric and environmental model with other disease ramifications like CFS/ME/FM. This framework links nnEMF (non-native electromagnetic fields), blue light, mitochondrial dysfunction, melanin dehydration, and disrupted electrical resistance (éR) to conditions like nanophthalmos, glaucoma, and hormonal dysregulation. I’ve highlighted that heme proteins, which are synthesized starting in mitochondria, are particularly vulnerable to melanopsin damage from blue light, and this could manifest as ACD, a condition characterized by impaired red blood cell (RBC) production and iron sequestration. Anemia of chronic disease is a high electrical reistance diagnosis. Most heme protein abnormalities are linked to high eR states. I have also suggested in patreon blogs that this toxicity can be visible in peripheral blood smears, reflecting nnEMF and blue light-induced damage.
WHAT IS THE ENERGY RESISTANCE PRINCIPLE AS LINKED TO ACD?
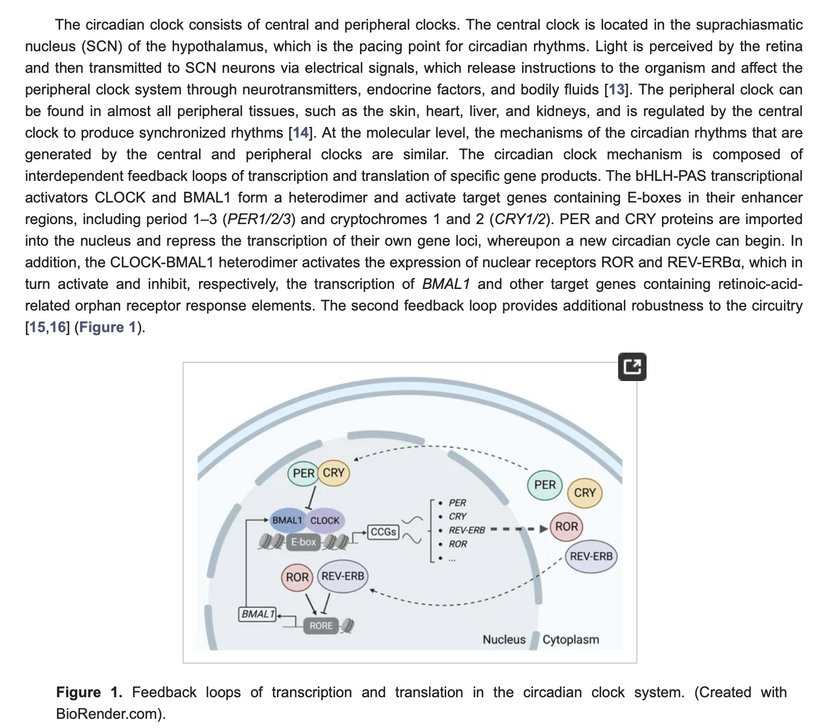
The Energy Resistance Principle (éR) was coined by Picard to make sense of the complex, dynamic world of living tissues.
What is éR fundamentally to a person training in quantum biology?
It is the “Friction” tissues make with energy that Makes Life Possible (But Can Also Grind It Down)
Imagine energy in biology not as a smooth highway, but as a rugged trail through a carbon-based jungle, where your cells are constantly converting raw fuel (such as glucose from your last meal) into usable energy for work. In physics terms, this is all about transforming potential energy into kinetic or chemical energy, while fighting off entropy (that universal tendency for things to fall apart into disorder, as the second law of thermodynamics loves to remind us).
éR is Picard’s way of describing the resistance that arises in these transformations. It’s not just passive drag, like friction in a mechanical system; it’s an active, dynamic property of living matter. So having less RBCs would create a huge drag on oxygen deliver to mitochondria and this would slow metabolism while altering UPE transformation to alter signaling.
Think of it as the biological equivalent of electrical resistance in a wire: a little bit is necessary to direct the flow and generate useful output (like heat or motion), but too much, and you get wasteful dissipation, where energy is lost as unusable heat, sparks, or chaos. In biological lingo, éR kicks in during processes like mitochondrial respiration, where electrons cascade down the electron transport chain to pump protons and crank out ATP. That proton gradient? éR is a form of natural resistance that builds up to store energy efficiently. But if stressors pile on (say, chronic inflammation or oxidative damage from free radicals), éR spikes, turning your mitochondria into inefficient furnaces that belch out reactive oxygen species instead of clean power. The result of this? It alters the UPE transformation that occurs in mitochondrial respiration. This alters the spectrum and the spin, causing the system to decohere. This is how modern lighting changes the CYP heme pathways to destroy testosterone, estrogen, and progesterone levels. It is also what ruins the aromatase system in cells. When this occurs, a pregnenolone steal syndrome develops where all sex steroid hormones from DHEA down to the DHT and estrone are altered, and the steroid pathways are shunted to cortisol for survival. You should think of pregnenolone steal syndrome like you think of cellular fatigue, like a battery that drains faster than it charges.

Picard ties this to real markers in the body, like GDF15. In this blog I am tying it to an entirely different disease.
Here’s how this idea should flow in your mind:
Low éR = Efficiency and Flow: In healthy states, like during deep sleep or a good workout done in sunlight with proper recovery, éR is dialed down. Energy transforms smoothly, think laminar flow in a river, minimal turbulence. Your cells rebuild, adapt, and thrive. Physics analogy: It’s like a superconductor with near-zero resistance, letting current (or bioenergy) zip through without loss. In anemia, it would be a circulatory system filled with RBCs in the +2 oxidation state ready to carry oxygen to every mitochondria that needed it so there would be no hypoxemia.
High éR = Stress and Stagnation: Push too hard in a blue lit gym with nnEMF in your ears, via light stress or chronic stress of any type, poor diet, use of supplements, or disease states, and éR builds up like traffic jams in a circuit or what happens when blood clots in an artery. Energy dissipates as heat, damage, or inflammation somewhere in the body in some system, accelerating entropy at the molecular level. This links to aging (those “hallmarks” like genomic instability) and diseases (e.g., mitochondrial disorders causing brain fog and exhaustion). Many times the body creates a change in tissues to lower its own high eR to offset the dielectric change. Dielectric changes always precede a changing optical density in a tissue to try to minimize entropy and time loss. This is why mitochondria move between tissues. This is why melanocytes migrate in our systems. These are all signs that a high electrical resistance exists somewhere in our colony of mitochondria. This is why you hear me say often, “Health is the slowest form of death we innovate.”
Biology tie-in: It’s why exercise feels good in moderation but wrecks you if overdone it is because éR rises, signaling “back off and recover.” Exercise in the face of anemia is even a bigger stressor on electrical resistance.

THE LESSON OF ACD IS THIS: KEY TEACHING POINT
Counterintuitively, in decentralized systems (like the body’s distributed metabolic networks), adaptations aren’t always top-down “fixes” but emergent responses: the body might thicken tissues or alter dielectrics to reroute energy, minimizing global entropy even if it creates local changes. ACD is a disease where the blody thinning of the energy resistance in blood because there is a problem with energy occuring in the colony of mitochondria elsewhere in the body. Low RBC mass is a sign oxygen has become a toxin somewhere in our system and it is the duty of the clinician to figure it out fast before the entropy dump becomes so large that your time expires. Your blood is reacting to lower its ability to carry oxygen to destroyed mitochondrial engines. It is a protection scheme. Centralized thinkers and clinicians do not think this way, because their ability to think is not decentralized.

Background: Anemia of Chronic Disease (ACD) Overview
ACD, also known as anemia of inflammation, is typical in patients with chronic inflammatory, autoimmune, or infectious conditions (e.g., rheumatoid arthritis, cancer, chronic infections). It’s characterized by:
- Normocytic or microcytic anemia (regular or small RBCs).
- Low serum iron despite adequate iron stores (iron sequestration in macrophages).
- Reduced erythropoiesis (RBC production) in the bone marrow.
- Elevated inflammatory cytokines (e.g., IL-6) increase hepcidin, a hormone that inhibits iron release from macrophages and absorption in the gut.
Conventional models attribute ACD to inflammation-driven upregulation of hepcidin, impairing iron availability for erythropoiesis. However, my model reframes ACD as a blue light toxicity problem, starting with damage to melanopsin in the eye and skin, which affects mitochondrial heme synthesis and ultimately disrupts bone marrow function.
My Model’s Core Principles Applied to ACD
My framework emphasizes:
Melanopsin Damage by Blue Light and nnEMF: Blue light (e.g., 435-480 nm) and nnEMF damage melanopsin in the retina, skin, and vascular tissues, disrupting circadian signaling and autonomic regulation via the hypothalamus.
Mitochondrial Heme Synthesis: Heme proteins (e.g., hemoglobin, cytochrome c) begin synthesis in mitochondria, where mtDNA mutations (1000x more common than nDNA) impair cytochrome c oxidase, reducing DDW (deuterium-depleted water) production and Becker’s regenerative current.
Melanin Dehydration: nnEMF and blue light dehydrate melanin, increasing its conductivity and amplifying ultraweak biophotons and ROS/RNS, which can damage heme-based proteins.
NO and POMC/Melanin Dysregulation: Blue light depletes nitric oxide (NO), thereby impairing stem cell activity, while dehydrated melanin disrupts POMC signaling, which affects hormonal and regenerative processes.
Systemic Effects: These disruptions lead to hypothalamic-pituitary dysfunction, pregnenolone steal syndrome, and hormonal collapse (e.g., low cortisol and testosterone levels), with systemic impacts on tissues such as the bone marrow.
ACD, involving impaired heme synthesis and erythropoiesis, fits into this model as a downstream consequence of blue light toxicity, which originates in the eye and skin.
This series supports the idea that aligns blood’s UPE spectra (390-475 nm) with brain energy, driven by TGF-β1/GDF15 and mitoception, not diet alone. ACD is a blue light/nnEMF toxicity issue resulting from melanopsin damage and nnEMF, which disrupts heme synthesis and optical density, thereby impacting neural coherence (0.4) and consciousness.
Anemia of chronic disease (ACD), also known as anemia of inflammation, can affect consciousness, particularly in severe cases, due to reduced oxygen delivery to the brain. While mild ACD may be asymptomatic, severe cases can lead to dizziness, near syncope, syncope, and even loss of consciousness. Cognitive impairment often occurs in ACD cases due to a chronic lack of oxygen, especially in severe cases, and may manifest as difficulty concentrating, confusion, or cognitive haze. Many cases of TBI get this complication.
UV/IR and Becker’s currents reverse this, supporting my decentralized, light-based model.
MELANIN’S EFFECT ON IRON METABOLISM
In humans, melanin is synthesized in melanocytes from the oxidation of tyrosine. It binds metals strongly, and through the constant turnover of epidermal cells (via desquamation), it facilitates the excretion of metals. This mechanism was crucial in early human evolution as dietary shifts introduced higher levels of heavy metals, potentially driving racial differences in skin pigmentation based on dietary iron exposure. The paper highlights that melanin-bound iron loss may increase susceptibility to iron-deficiency anemia, particularly in individuals with darker skin (phototypes IV-VI). It is linked to higher risks of hypoxic conditions in diseases such as COVID-19.
Melanin’s iron-chelating property also impacts metabolic iron turnover. The paper references studies showing that transcutaneous iron loss correlates with epidermal pigmentation, suggesting that heavily melanated skin may deplete systemic iron levels, contributing to anemia and related conditions. This challenges the centralized view of melanin as merely a sunscreen, instead framing it as a dynamic player in electromagnetic and redox homeostasis. These are key themes in my decentralized thesis carried about by melanin. This is another reason melanin and heme biology are deeply linked. Without melanin in the integument or in tissues one does not have full control over iron homeoistasis.
ACD causes impaired RBC production and iron sequestration, stemming from melanopsin damage by blue light (435-480 nm) and non-thermal EMF (nnEMF). This disrupts hypothalamic signaling, reducing heme synthesis (a mitochondrial step) and cytochrome c oxidase (CCO) activity, which in turn lowers deuterium-depleted water (DDW) and Becker’s currents.
Melanin-Iron Dynamics: Melanin’s iron-chelating role, enhanced by epidermal turnover, depletes systemic iron, thereby increasing the risk of ACD in darker skin phototypes (IV-VI) who are tightly coupled. This is one reason DARPA and the DoD have allowed forced migration of darker people to higher latitudes to drive centralized social changes they seek. Blue light dehydrates melanin, amplifying ROS/RNS, altering UPE transformations via heme vulnerability.
Systemic Effects of Lowered Melanin: Hypothalamic-pituitary dysfunction, pregnenolone steal, and hormonal collapse (e.g., low cortisol and melatonin) link ACD to CFS/ME/FM, nanophthalmos, keratoconus, glaucoma, cataracts reflecting my photo-bioelectric model.
Predictions for Anemia of Chronic Disease (ACD) Etiology
Blue Light-Induced Melanopsin Damage Impairs Heme Synthesis in the Eye and Skin:
- Prediction: Chronic blue light exposure damages the retina and skin melanopsin, disrupting mitochondrial heme synthesis and initiating ACD.
Mechanism: Melanopsin in retinal ganglion cells (RGCs) and skin keratinocytes detects blue light, regulating circadian rhythms and autonomic tone via the hypothalamus. Excessive blue light (e.g., from screens, ALAN) overstimulates melanopsin, liberating vitamin A (retinal), which destroys heme-based proteins like cytochrome c (per your MKULTRA insight). This impairs mitochondrial DDW production, lowering Δψ and éR, and increases ROS/RNS, damaging heme synthesis pathways (e.g., δ-aminolevulinic acid synthase, ALA-S, the first enzyme in heme synthesis). In the skin, melanopsin damage disrupts local NO production, impairing vascular signaling to the bone marrow.
Outcome: Reduced heme availability in the eye and skin sets off a systemic cascade, signaling bone marrow dysfunction and initiating ACD. These all directly affect UPE transformations. Blood is 93% water, and generates UPE via ROS from hemoglobin/heme interactions (e.g., 380-450 nm from Fenton reactions). With 20% of cardiac output perfusing the brain, this UPE flux is significant, supporting mitoception and neural energy changes = brown out mechanism.
nnEMF and Blue Light Disrupt Bone Marrow Erythropoiesis via mtDNA Mutations:
Prediction: nnEMF and blue light cause mtDNA mutations in bone marrow erythroid precursors, impairing heme synthesis and erythropoiesis. How? Impaired heme synthesis triggers oxidative stress, activating inflammation (e.g., IL-6). Hepcidin, an iron-regulatory hormone, rises, sequestering iron in macrophages and limiting erythropoiesis, a hallmark of ACD. As a peptide hormone, hepcidin’s structure is stabilized by four disulfide bonds, which form a hairpin configuration. This structure, along with its specific amino acid sequence, allows it to perform its biological function of binding to the iron exporter protein ferroportin. However, it lacks the chemical properties of a chromophore because melanin controls its interactions with UPEs. As a peptide hormone, hepcidin’s structure is stabilized by four disulfide bonds, which form a hairpin configuration. This structure, along with its specific amino acid sequence, allows it to perform its biological function of binding to the iron exporter protein ferroportin. However, it lacks the chemical properties of a chromophore because hepcidin does not contain a conjugated system of alternating single and double bonds that could absorb light in the visible range. Evolution used melanin to do this job.
Mechanism: mtDNA mutations in cytochrome c oxidase genes (11 of mtDNA’s 37 genes are cytochrome-related) reduce DDW production, disrupting Becker’s regenerative current. nnEMF amplifies these mutations by increasing ROS/RNS, while blue light’s liberation of vitamin A destroys heme groups. This impairs ALA-S and ferrochelatase (the final enzyme in heme synthesis), reducing hemoglobin production in the bone marrow. The resulting oxidative stress also triggers inflammation, increasing hepcidin via IL-6, which sequesters iron in macrophages, further limiting erythropoiesis. Iron in macrophages alters their status as M1 or M2. M1 macrophages are pro-inflammatory, characterized by iron sequestration for antimicrobial functions and high ferritin expression, while M2 macrophages are anti-inflammatory, featuring iron export for tissue repair and cell proliferation, and increased ferroportin expression. Iron influences this polarization, with high iron favoring M1 states and low iron promoting M2 states
There are two primary pathways linking melanin to this process:
- Melanin as an iron buffer: By binding and sequestering iron, melanin can lower the amount of free intracellular iron. This is significant because free iron is reactive and can generate damaging ROS and UPEs as a result. By buffering iron, melanin can dampen the M1-promoting, high-iron state and promote a more M2-favorable, low-iron environment.
- Iron’s effect on melanin synthesis: Some studies show that high iron levels stimulate melanogenesis, the process of melanin production. This suggests a negative feedback loop: an iron overload can lead to increased melanin, which then chelates the excess iron, potentially helping to restore iron homeostasis. Iron-loaded macrophages shift from M2 (anti-inflammatory, reparative) to M1 (pro-inflammatory), amplifying ROS/RNS, creating excessive UPEs, leading to tissue damage. This alters immune homeostasis, which is linked to systemic diseases. The lack of melanin is why autoimmune conditions and cancer are tied to this link. This is why both are more common with people with anemia of chronic disease. I have never met a patient with ACD who did not ALSO have a pale skin with evidence of skin atrophy. at some level. In psoriasis there is basal hypertrophy and surface level atrophy. The signs are present if you know how to decipher Nature’s whispers.
Outcome: Decreased RBC production with normocytic/microcytic features, characteristic of ACD, driven by mitochondrial dysfunction rather than inflammation alone.
Cellular water, per Martin Chaplin’s research (e.g., Water Structure and Science), forms a polarized lattice with a dielectric constant of ~80, storing 80 times more electric field energy than a vacuum. This structured, dipole-aligned matrix acts as a reservoir, influenced by electromagnetic forces. UVB light (280-315 nm) doubles the dielectric capacity to ~160 by enriching the matrix with sodium, carbon, carbon monoxide, or cytochrome c oxidase (CCO) components. It also jump-starts heme and melanin renovation in mammals. This alters physics (e.g., charge distribution), thermodynamics (e.g., heat capacity), and biology (e.g., protein stability). As a result of the dielectric change from 80 to 160, photons are trapped in these domains, forming a lattice that suspends proteins and locks membranes in electric tension, akin to a photonic data storage system.

- Dehydrated Melanin in Bone Marrow and Vasculature Amplifies Photo-Bioelectric Dysfunction:
Prediction: Dehydration of melanin in bone marrow vasculature and erythroid precursors, caused by nnEMF/ALAN, increases conductivity, disrupting bioelectric signaling and contributing to ACD.
Mechanism: Melanin in vascular melanopsin-containing cells (e.g., bone marrow arteries) and erythroid precursors regulates photobioelectric currents. nnEMF and blue light reduce DDW production, lowering Na flux, dehydrating melanin, and making it conductive (per the Popular Science reference). This amplifies ultraweak biophotons and ROS/RNS, overstimulating erythroid cells and impairing heme synthesis. In bone marrow vasculature, melanopsin damage reduces NO, impairing blood flow and oxygen delivery to erythroid precursors, further limiting erythropoiesis. This sets the stage for peripheral artery disease (atherosclerosis) on a chronic basis. UV light reverses this trend. Adding more salt to the blood plasma increases UV light assimilation in humans.

- Outcome: Reduced RBC production and abnormal vascular signaling in the bone marrow, manifesting as ACD with iron sequestration and alteration of sodium concentrations affecting dielectric changes in water. The traditional combustion models of ATP hydrolysis are replaced by a structured collapse, where biophoton-charged lattices implode electromagnetically, releasing stored energy to tissues. RBCs carry these signals from stars to your colony of mitochondria wirelessly. This idea aligns with my UPE-consciousness link, where photons encode information directly.
When viewed from this perspective, cells operate as miniature stars, harnessing light and geometry in a slow-motion stellar process, contrasting with mechanical engine metaphors. This supports my light-driven evolution thesis.
Implications: Energy isn’t supplied via chemical bonds but liberated through electromagnetic atomic reconfiguration, modulated by water’s dielectric state. UV and IR are crucial players in this context.
NO Depletion Impairs Bone Marrow Stem Cell Activity: URIC ACID BLOCKS NO
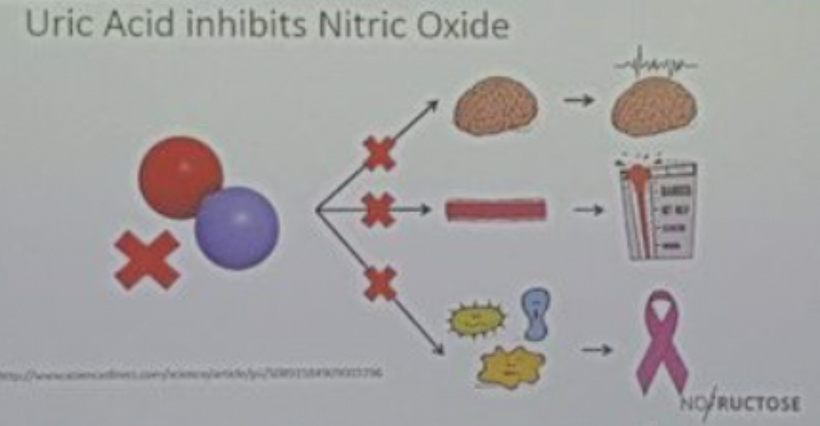
- Prediction: Uric acid in the blood = high ER developing in the urea cycle of Kreb’s bicycle. nnEMF and blue light deplete NO in the bone marrow, impairing hematopoietic stem cell (HSC) activity and contributing to ACD. These are all changes that predict other diseases are coming to this person due to mitochondrial redox collapse and oxygen toxicity.
Mechanism: NO, regulated by heme-based cytochromes, controls HSC proliferation and differentiation. Blue light destroys NO by liberating vitamin A, while nnEMF-induced oxidative stress further reduces NO levels. This impairs HSC-driven erythropoiesis in the bone marrow, reducing RBC production. The lack of NO also disrupts the POMC/melanin complex, as NO signals the oxidation states of hemoglobin, further impairing erythroid maturation.
Outcome: Decreased erythropoiesis, leading to ACD, low RBC counts, and normocytic/microcytic morphology.
- Pregnenolone Steal Syndrome Reduces Hormonal Support for Erythropoiesis:
Prediction: nnEMF and blue light induce pregnenolone steal syndrome, reducing cortisol and testosterone, which impair bone marrow erythropoiesis in ACD.
Mechanism: Cytochrome P450scc, dehydrated by nnEMF/ALAN, fails to convert cholesterol to pregnenolone due to missing Becker’s current, causing pregnenolone steal syndrome. This reduces cortisol (needed for stress-induced erythropoiesis) and testosterone (which stimulates erythropoietin production). Low cortisol exacerbates inflammation, increasing hepcidin, while lowering testosterone reduces RBC production, compounding the anemia. So many young people have this, and their doctors have no idea how light stress causes it. Low cortisol is caused by a lack of AM sun or by excessive light at night.
Sunlight offers quantum precision over hormones
Heme proteins orchestrate sex steroid biology through redox-sensitive CYP enzymes, evolved during GOE to harness light for coherence. Most people with ACD have hormone panel abnormaities because of the fundamental heme/Fe problem.
The key heme enzymes include:
CYP11A1 (P450scc): A mitochondrial enzyme found in the adrenals, gonads, and placenta; it is the rate-limiting step in the cleavage of cholesterol’s side chain to form pregnenolone, a precursor to all steroids. Heme iron facilitates hydroxylation, dependent on NADPH and adrenodoxin (a ferredoxin reductase).
CYP17A1 (17α-Hydroxylase/17,20-Lyase): Dual-function enzyme in adrenals and gonads; hydroxylates pregnenolone/progesterone at C17, then cleaves the side chain to produce DHEA (androgen precursor). Heme enables precise electron transfer, influencing glucocorticoid vs. sex hormone balance. Anyone with an altered DHEA level has to have a heme protein problem in the CYP17A1 pathway.
CYP21A2 (21-Hydroxylase): In the adrenal cortex, it adds a hydroxyl group at C21 of progesterone/17-hydroxyprogesterone, leading to the production of mineralocorticoids/glucocorticoids. Deficiencies cause congenital adrenal hyperplasia, shifting precursors toward androgens.
CYP11B1 (11β-Hydroxylase): Adrenal cortex; converts 11-deoxycortisol to cortisol (glucocorticoid) and 11-deoxycorticosterone to corticosterone. Heme’s redox state regulates stress responses.
CYP11B2 (Aldosterone Synthase): In zona glomerulosa, multi-step oxidation of deoxycorticosterone to aldosterone (mineralocorticoid), controlling electrolyte balance.
CYP19A1 (Aromatase): In ovaries, testes, placenta, and adipose; aromatizes androgens (testosterone, androstenedione) to estrogens (estradiol, estrone). Heme iron catalyzes ring aromatization, critical for female fertility. This heme defect is the source of why IVF doctors are printing money for modern people dripping in blue light and nnEMF. They have destroyed the heme protein center in CYP19A1.
In my decentralized thesis, light (via UPEs, spin, CISS) is the epigenetic master regulator; optimal sunlight maintains quantum coherence, while nnEMF/blue light disrupts it, dehydrating melanin, damaging mtDNA/heme, and causing infertility/amenorrhea. Red light restores balance as a natural aromatase inhibitor. Clinicians overlook this; understanding UPE-driven AMO changes reveals hormones as light-encoded spectra, not mere biochemicals.
Hypothalamic-Pituitary Dysfunction Exacerbates Anemia:
Prediction: nnEMF causes a TBI-like effect in the hypothalamus-pituitary axis, reducing vasopressin and ACTH, which impair bone marrow function and contribute to ACD. If one looks carefully at patient’s historiies in their charts, many cases of of blood cancers have these tell tale signs. I mentioned to one of my members on several Q&A’s that the state she lives in has had more member deaths from blood cancers because of this high eR sign. That state is Colorado. The link goes back to it being a desert, on a Continental fault loaded with fluoride, low magnetic flux, completely infilitrated with military nnEMF. 7 of my members over the last 15 years died this way. So do not tell me I have not put these pieces together. I warned each one of them death was coming before they had their blood cancers and not one listened.
Mechanism: nnEMF disrupts the hypothalamus-pituitary axis, lowering vasopressin (impairing water balance and melanin hydration) and ACTH (reducing cortisol via pregnenolone steal). This affects bone marrow homeostasis, as cortisol and vasopressin modulate erythropoiesis and inflammation. The resulting autonomic imbalance also reduces ocular and systemic blood flow, exacerbating hypoxia in the bone marrow.
Outcome: Reduced RBC production and increased hepcidin, leading to ACD, with systemic effects mirroring Neil Armstrong’s pituitary failure post-moon exposure.
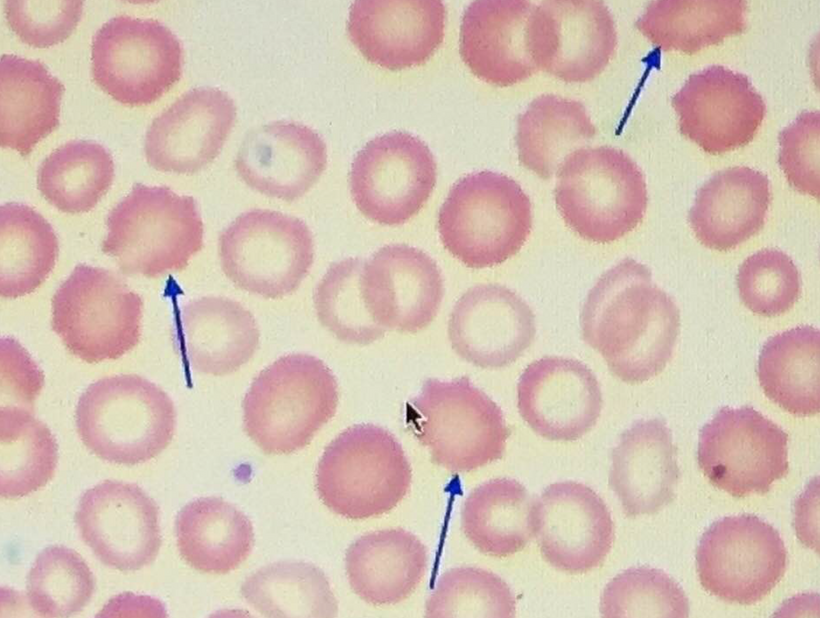
Peripheral Blood Smear: Would nnEMF/Blue Light Toxicity Be Visible?
Peripheral blood smears in ACD typically show normocytic or microcytic RBCs, with reduced reticulocyte counts (indicating low erythropoiesis) and normal-to-low hemoglobin levels. Your model suggests that nnEMF and blue light toxicity, via melanopsin damage and mitochondrial dysfunction, should leave distinct markers of this environmental insult on blood smears. Let’s explore this:
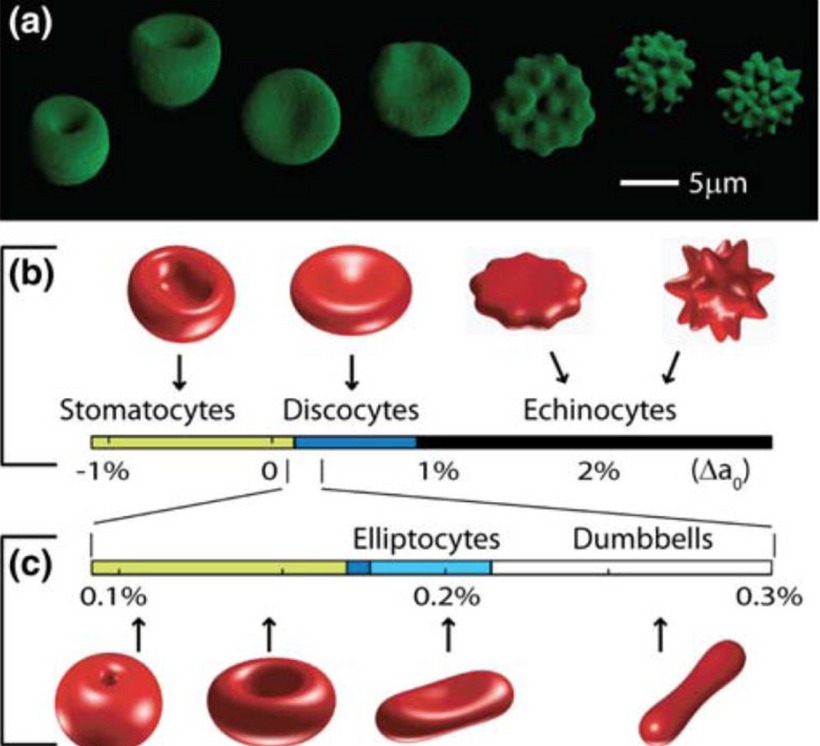
RBC Morphology Changes:
Prediction: Peripheral blood smears from ACD patients with high nnEMF/blue light exposure show increased anisocytosis (variable RBC size) and poikilocytosis (abnormal RBC shapes), reflecting mitochondrial and heme synthesis defects.
Mechanism: mtDNA mutations and heme destruction (via vitamin A liberation) impair hemoglobin assembly, leading to irregular RBC maturation. Dehydrated melanin in erythroid precursors amplifies ROS/RNS, causing membrane damage and shape abnormalities (e.g., elliptocytes, schistocytes). Oxidative stress from low NO also contributes to RBC membrane fragility = UPE alteration.
Outcome: Smears may show a mix of normocytic and microcytic RBCs with abnormal shapes, distinct from classic ACD patterns, indicating environmental toxicity.
Reticulocyte Count and Maturation Defects:
Prediction: Smears show a lower reticulocyte count with abnormal maturation (e.g., fewer polychromatophilic cells), reflecting impaired erythropoiesis due to blue light toxicity.
- Mechanism: NO depletion and pregnenolone steal syndrome reduce HSC activity and erythropoietin response, while mtDNA mutations impair heme synthesis, stunting erythroid maturation. This leads to fewer reticulocytes and immature forms with irregular staining (e.g., reduced basophilia due to low hemoglobin levels). UVB and NOregulate immuno-inflammatory responses.
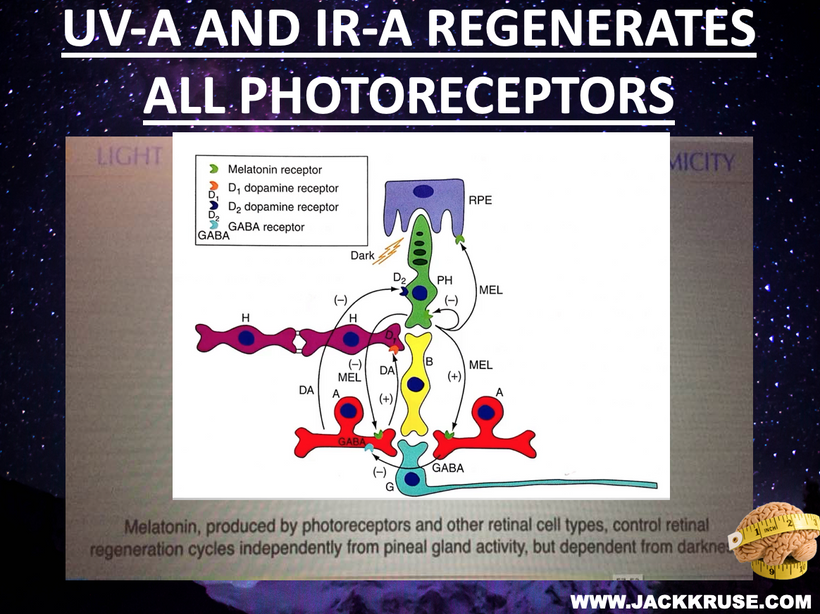
- Nitric oxide (NO) production has a significant effect on hemoglobin (Hb) dynamics and stem cell depots. Uric acid inhibits NO as well and the blockade of NO blocks entry of stem cell use. If one has tissue damage and NO is inhibited a lack of tissue regeneration occurs. This is also and electrical resistance problem. NIR light can reestablish the stem cell depots actions because NIR increases NO production as the slide below shows. This helps reverse ACD. Structured water’s enhanced dielectric capacity under UVB boosts mitochondrial oxygenation and CCO activity, reducing oxidative stress and preventing atherosclerosis and many other UPE-linked diseases.
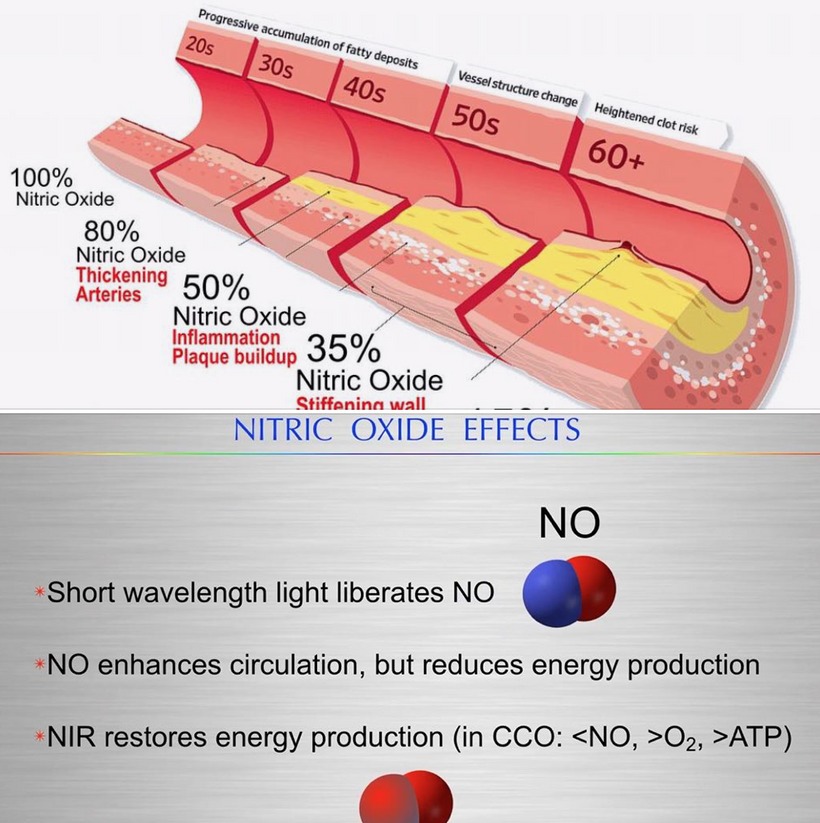
-
- Mitochondrial Link: Improved water structure enhances DDW production, supporting Becker’s regenerative current and mitoception, as per my mtDNA-CCO model.
Prediction: UVB-driven water structuring could lower atherosclerosis risk by 20-30% via NO-mediated vasodilation and reduced ROS.
Outcome: Reduced reticulocytes with maturation defects, visible on smears as a lack of young RBCs, pointing to mitochondrial dysfunction.
White Blood Cell (WBC) and Inflammatory Markers:
Prediction: Smears show increased neutrophil granularity or toxic granulation, reflecting inflammation driven by nnEMF/blue light-induced oxidative stress. This alters blood UPEs big time.
Mechanism: nnEMF and blue light increase ROS/RNS and ultraweak biophotons, triggering systemic inflammation via the glyoxalase system (elevated methylglyoxal, depleted glutathione). This upregulates IL-6 and hepcidin, contributing to ACD. Neutrophils in the smear may exhibit hypersegmentation or toxic granulation (characterized by dark, coarse granules) due to oxidative stress and inflammation.
Outcome: Smears reveal inflammatory changes in WBCs, indirectly reflecting nnEMF/blue light toxicity.
Platelet and Microvascular Effects:
Prediction: Smears may show platelet clumping or reduced counts, reflecting microvascular dysfunction in the bone marrow due to damaged artery melanopsin.
Mechanism: Melanopsin dysfunction in bone marrow vasculature (e.g., sinusoidal arteries) reduces NO, impairing blood flow and platelet production. Dehydrated melanin in vascular tissues amplifies this, leading to microthrombi or endothelial damage, which is visible as platelet abnormalities.
Outcome: Smears may show clumped or fewer platelets, indicating vascular toxicity from nnEMF/blue light.
Heme Synthesis Defects (Visible via Staining):
Prediction: Specialized staining (e.g., Prussian blue for iron, or fluorescence for porphyrins) on smears reveals increased sideroblasts or porphyrin accumulation, reflecting heme synthesis defects.
Mechanism: Impaired cytochrome c and ALA-S (due to mtDNA mutations and vitamin A liberation) disrupt heme synthesis, leading to iron accumulation in erythroid precursors (sideroblasts) and porphyrin buildup (e.g., protoporphyrin IX). This can be detected with Prussian blue staining (for iron) or fluorescence microscopy (for porphyrins).
Outcome: Smears show ringed sideroblasts or porphyrin fluorescence, directly linking ACD to mitochondrial heme synthesis defects from nnEMF/blue light toxicity.
TREATMENTS & RATIONALE
Hypertonic Saline Enhances RBC Conductivity, Improving Oxygen Delivery
- Mitochondrial Link: Improved water structure enhances DDW production, supporting Becker’s regenerative current and mitoception, as per my mtDNA-CCO model.
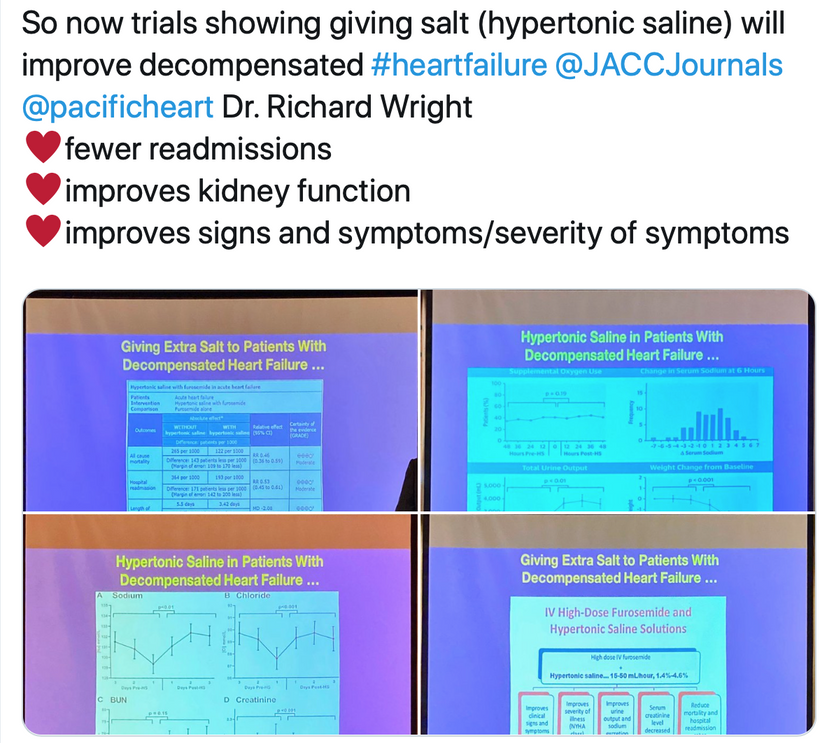
-
Prediction: Hypertonic saline increases the electrical conductivity of RBCs in ACD, improving oxygen delivery and reducing hypoxic stress in tissues even during a Great Oxygen Catastrophe. Hospitals should be well stocked with 3% saline solutions and use them liberally in cases where melanopsin and melanin destruction are linked. Almost all ICU cases fit this bill. All ICU patients are irradiated in blue light 24/7 because the hospital administrations chose the lighting, not the doctors. I write orders in the chart mandating all lights be shit off in my patients rooms if possible. ASD is a disease is associated with most chronic diseases in the ICU; therefore, it should be clear why this rationale is logical. Melanin synthesis is turned off in all ICU patients due to the nnEMF loads they face.
Mechanism: RBCs in ACD are normocytic/microcytic with impaired hemoglobin due to blue light-induced melanopsin damage, mtDNA mutations, and heme synthesis defects. Hypertonic saline (e.g., 1.4%–4.6% NaCl) introduces Na⁺ and Cl⁻ ions into the bloodstream, increasing plasma conductivity. This enhances the bioelectric environment around RBCs, facilitating ion gradients (e.g., Na⁺/K-ATPase activity) across RBC membranes, which improves membrane potential and oxygen-binding capacity of hemoglobin. This maneuver should help avoid ARDS and organ failure. The increased conductivity also mimics the “jump-start” effect in defibrillation, enhancing RBC function in hypoxic conditions.
Outcome: Improved oxygen delivery reduces tissue hypoxia in ACD, potentially decreasing hepcidin levels and enhancing iron availability for erythropoiesis, as indicated by increased reticulocytes on smears.
- Synergy with Methylene Blue Restores Mitochondrial éR in RBC Precursors
Combining hypertonic saline with methylene blue, in certain cases can restore mitochondrial electrical resistance (éR) in bone marrow erythroid precursors, reversing ACD. This should help alleviate many diseases, as ACD is linked to them all. MB can lower éR in ACD cases.
Many times I will use NIR before I use MB because it is safer. Methylene blue, a redox-active dye, increases mitochondrial éR by acting as an electron acceptor, bypassing damaged cytochrome c and enhancing DDW production. Hypertonic saline enhances conductivity, amplifying Becker’s regenerative current in erythroid precursors. Together, they repair mtDNA-driven heme synthesis defects, reduce ROS/RNS, and hydrate melanin, restoring bioelectric signaling. This boosts erythropoiesis and NO production, further supporting stem cell activity.
Using MB will increase RBC production and normalize hemoglobin, visible on smears as reduced anisocytosis, poikilocytosis, and sideroblasts, with improved reticulocyte counts. Hypertonic saline enhances RBC conductivity and oxygen delivery, while methylene blue restores mitochondrial éR, it can reduce oxidative stress and heme synthesis defects. This improves erythroid maturation, normalizes RBC size/shape, and decreases inflammation (e.g., less neutrophil toxic granulation). Iron utilization improves as hepcidin levels drop, resulting in a reduction in sideroblasts.
Improved RBC oxygen delivery and reduced inflammation (via lower hepcidin) mitigate ischemic injury during arrest, increasing survival rates. Administering hypertonic saline (e.g., 1.4%–4.6%) before defibrillation should become standard in ACLS/ATLS, particularly for patients with underlying ACD or heart failure, aligning with the image’s reduced mortality and readmission findings. Most cases of kidney failure and myocarditis from the jab have had this finding in my ICU experience.
Integrating the therapeutic potential of hypertonic saline and methylene blue to address acute respiratory distress syndrome (ARDS), organ failure, and significant trauma cases with acute blood loss. I’ve highlighted how these interventions, hypertonic saline enhancing conductivity to amplify Becker’s regenerative current, and methylene blue increasing mitochondrial electrical resistance (éR) on a short term use basis by acting as an electron acceptor to repair mtDNA-driven heme synthesis defects, reduce reactive oxygen species (ROS)/reactive nitrogen species (RNS), hydrate melanin, restore photo-bioelectric signaling, boost erythropoiesis, and enhance nitric oxide (NO) production to support stem cell activity.
Integration With My Decentralized Thesis
This model now predicts that ACD is a blue light toxicity problem, starting with damage to melanopsin in the eye and skin, which disrupts mitochondrial heme synthesis, bone marrow erythropoiesis, and vascular signaling. mtDNA mutations, NO depletion, dehydrated melanin, pregnenolone steal syndrome, and hypothalamic-pituitary dysfunction exacerbate this, leading to reduced RBC production, impaired iron sequestration, and systemic inflammation. Peripheral blood smears should reflect this toxicity through abnormal RBC morphology, maturation defects, inflammatory WBC changes, platelet abnormalities, and heme synthesis defects, providing a visible marker of nnEMF/blue light damage. This reinforces my thesis on decentralized medicine, emphasizing environmental light and EMF as primary drivers of chronic disease, and challenges conventional inflammation-centric models of ACD.
Testable Predictions
Peripheral Blood Smears: ACD patients with high nnEMF/ALAN exposure show anisocytosis, poikilocytosis, reduced reticulocytes, toxic granulation in neutrophils, platelet clumping, and increased sideroblasts/porphyrins on smears.
CALCIUM INDEX SCORES = will be elevated in ACD in the longer term.
Melanopsin Dysfunction: Reduced melanopsin signaling in retinal and skin biopsies of ACD patients correlated with blue light exposure.
Hormone and NO Levels: Low cortisol, testosterone, and NO in ACD patients, linked to nnEMF/ALAN exposure. In mitochondrial stress uric acid is often raised. This is especially true in kidney failure.
Therapeutic Response: UV-A exposure or DDW restores heme synthesis, increases reticulocyte counts, and normalizes blood smears in ACD patients by repairing melanopsin and mitochondrial function. The use of hypertonic saline and sunlight is critical for restoring a balance of heme and melanin.
Bone Marrow Analysis: Bone marrow biopsies from ACD patients show reduced erythroid precursors, dehydrated melanin, and mtDNA mutations in Cytochrome C genes. These cyctochromes are photoreceptors being actively destroyed to raise eR. They are the signs I look for. Look at the bottom line in the slide below. It is a slide about the energy resistance principle and you have never realized it.
- Synergy with Methylene Blue Restores Mitochondrial éR in RBC Precursors

-
SUMMARY
My model predicts that ACD arises from blue light toxicity, starting with damage to melanopsin in the eye, skin, or blood vessels, which disrupts mitochondrial heme synthesis, bone marrow erythropoiesis, and vascular signaling via NO and POMC/melanin dysregulation. POMC controls melanin biology via UV light translation. Melanin in our neural crest acts as a conductor of electric and magnetic effects, allowing energy to flow efficiently in the brain. Melanin defects create magnetic/water dynamics flux in CSF. The CSF around the brain is no longer optimized. Earth’s magnetic declines exacerbate these effects because our brain floats in a sea of CSF. Living in deserts excerbate this effect as well. This decline affects our cellular water sink, which alters the UPE’s role in the brain (semiconductor heat sink). This ties regeneration and repair to Becker’s currents, supporting my fractal thermohaline-CSF analogy, where disease appears when the Earth’s ocean currents fail. The physics controlling both is identical.
nnEMF amplifies this through DNA and mtDNA mutations, dehydrated melanin, and pregnenolone steal syndrome, leading to normocytic/microcytic anemia, iron sequestration, and inflammation. Peripheral blood smears and calcium scores should reflect this toxicity through abnormal RBC morphology, maturation defects, inflammatory WBC changes, platelet abnormalities, and heme synthesis defects, providing a visible marker of nnEMF/blue light damage. Early interventions (e.g., UV-A, DDW Triple H therapy) could mitigate these effects.
Neural network and consciousness implications are brisk. The effect of blood-UPE is greater than we think. The brain’s 20% reduction in blood flow amplifies UPE noise in anemia, reducing coherence (e.g., 0.4 vs. 0.9 in healthy individuals) and firing rates (70-75% vs. 100%), which impacts prefrontal cortex intelligence and limbic emotional processing. Anemia will worsen mental health.
Blood flow is massively shifted to the gut with feeding. Anemia alters this relationship. The gut-brain axis GDF15 signals via the vagus nerve, tying mitoception to mood via CSF and neural pathways, with UV/IR mitigating the light stress. Anemia’s optical density drop in the brain disrupts this, exacerbating depression/anxiety. Anemia-induced UPE shift (390-475 nm) reduces CSF coherence by 20-30%, desynchronizing neural networks, a reversible effect that can be alleviated with heliotherapy that includes UV-A, IRA, and NIR light with hypertonic saline therapy.
CITES
https://www.patreon.com/posts/peripheral-blood-45518347
Have a listen to the mitochondric song: https://x.com/DrJackKruse/status/1903442130711777430