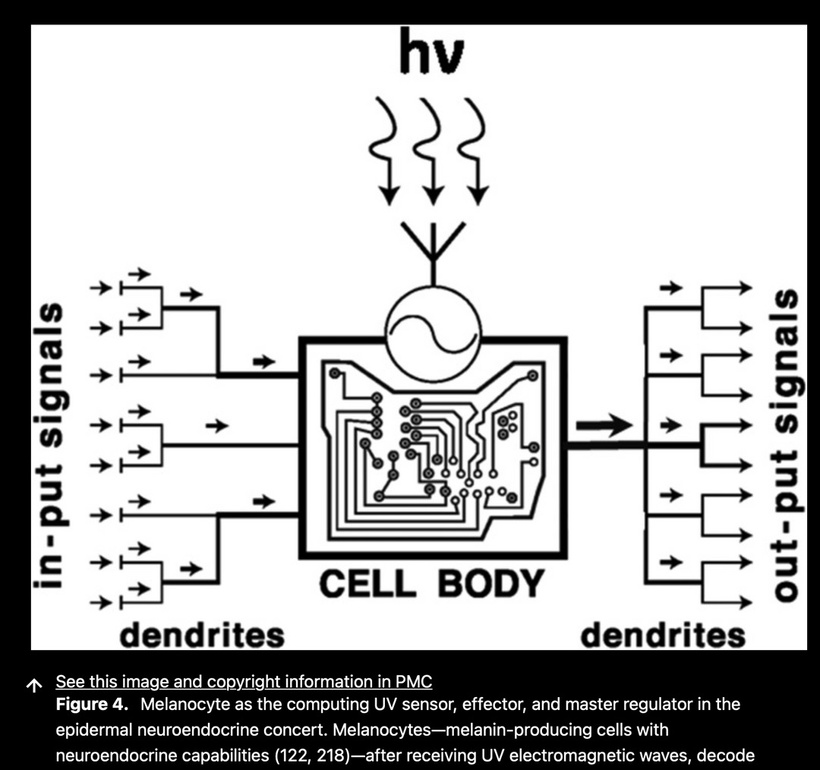
WHY ARE EYE DISEASES MORE COMMON TODAY?
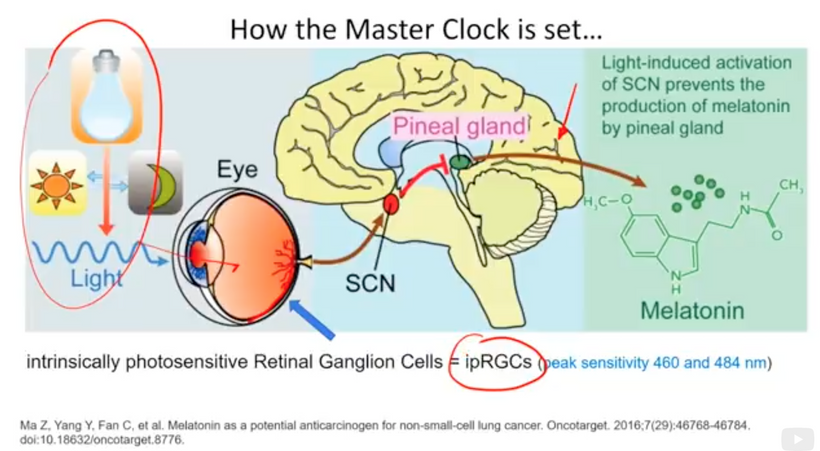
The neurosensory retina is a layered tissue that lines the back of the eye, communicating with the brain via the optic nerve. Blood is supplied to the neurosensory retina by retinal blood vessels originating from the central retinal artery. Transport across retinal blood vessels is limited by endothelial tight junctions, which constitute the inner blood–retinal barrier. Allan Frey’s work with DARPA in the 1960s demonstrates that non-thermal electromagnetic fields (nnEMF) open this barrier and make the eye more susceptible to damage when present in our local environment.
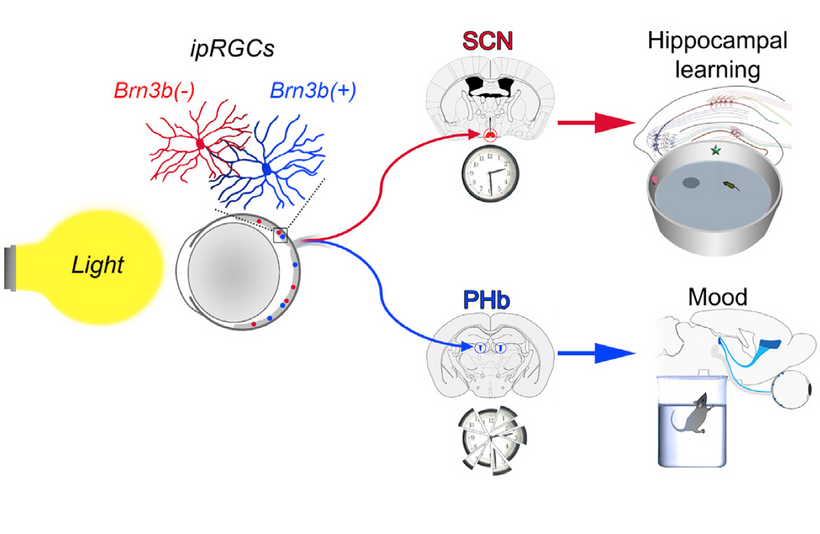
We forget that the optic nerve is comprised of ganglion cell axons, the “output neurons” of the retina. The retina is composed of three layers of cell bodies and two plexiform layers of synapses, which are clearly defined on histological sections. The innermost layer is the ganglion cell layer. The inner nuclear layer, featuring the cell bodies of bipolar, amacrine, and horizontal cells, is sandwiched by an internal plexiform layer and ganglion cell layer on one side, and outer plexiform layer and outer nuclear layer on the other. The outer nuclear layer contains the cell bodies of most photoreceptors, including the rods and cones. A third class of photoreceptor, intrinsically photoreceptive ganglion cells (melanopsin blue light detectors), is situated in the ganglion cell layer. All photoreceptors depend on a vitamin A-derived chromophore to detect light and adequate dopamine and melatonin levels to regenerate them in sunlight. The chromophore associated with light-transducing proteins, opsins, undergoes photoisomerization during light transduction. Expended and spent melanopsin or neuropsin chromophores and their associated retinal then enter the visual cycle, a sequence of reactions that facilitates regeneration and re-association with opsins for further transduction. If regeneration pathways are blocked, the free Vitamin A (retinal)becomes a wrecking ball for the human retina.
THEY ARE BLINDING US WITH BLUE WHILE STOPPING UV-IR REGENERATION Rx.
Carnot’s theorem, formulated by Sadi Carnot in 1824, establishes the maximum efficiency of a heat engine, defined as the ratio of the temperature difference between the heat source (e.g., the engine’s interior) and the cold sink (e.g., the surroundings) to the absolute temperature of the heat source. In biological terms, as you’ve suggested, mitochondria can be viewed as “hydrogen heat engines” that produce ATP (energy) by maintaining a proton gradient across their inner membrane, effectively creating a temperature and electrochemical gradient. Cooling can tan your interior and your exterior and centralized science ignores it.

Application to Mitochondria: In retinal cells, which have high metabolic demands due to constant phototransduction, mitochondria generate heat and energy. According to Carnot’s principle, the efficiency of these mitochondrial “engines” increases with a greater temperature differential between the mitochondrial matrix (heat source) and the cellular environment (cold sink). Cold thermogenesis, which involves exposing the eye to cold, enhances this gradient by lowering the external temperature, forcing mitochondria to work harder and produce more heat, thereby improving efficiency and resilience.
Impact on Eye Health: Under indoor conditions with stable temperatures (e.g., thermostat-controlled environments), this gradient diminishes, leading to mitochondrial atrophy. This reduces the retina’s ability to repair damage from blue light hazard, where reactive oxygen species (ROS) from all-trans-retinal accumulation overwhelm cellular defenses. My chronic point about “chronic widespread mitochondrial colony failure” due to lack of seasonal variation aligns here, because without cold stress, mitochondrial efficiency drops, exacerbating retinal degeneration.

Haplotypes and Susceptibility to Blue Light Hazard
Haplotypes, which are specific combinations of genetic variants inherited together, can influence mitochondrial function, melanin biology, and photoreceptor health, potentially making certain populations more vulnerable to blue light damage.
Mitochondrial Haplogroups: Human mitochondrial DNA (mtDNA) is organized into haplogroups (e.g., H, U, J, T), which vary by geographic origin and affect metabolic efficiency. For instance:
Haplogroup H (common in Europe) is associated with higher basal metabolic rates but may be less adaptable to oxidative stress, potentially increasing susceptibility to blue light-induced ROS in the retina.
- Haplogroup J (prevalent in the Near East and Caucasus) shows enhanced thermogenic capacity and cold tolerance, possibly offering better mitochondrial resilience against blue light damage.
Studies (e.g., Mitochondrion, 2015) suggest that haplogroups with lower electron transport chain efficiency (e.g., some African haplogroups like L0) might accumulate more ROS under blue light stress, heightening retinal risk. Their skin and RPE also have more melanin, so they need much higher amounts of sunlight to tap into their regeneration programs. This is why African Americans have such high rates of cataracts, diabetes, and retinal diseases when they move to America, where the technocracy destroys their retinal biology.

Melanin and Photoprotection: Haplotypes also influence melanin production via POMC and MC1R gene variants. Lighter-skinned individuals (e.g., those with haplogroup H) have less melanin in the retinal pigment epithelium (RPE), which reduces photoprotection against blue light. Darker-skinned individuals (e.g., haplogroup L) with higher melanin levels are more resistant, as melanin scavenges ROS and conducts bioelectric signals (per Becker’s work). This increases the signal-to-noise ratio.

Heat Production Link: Mitochondria in haplotypes with lower thermogenic capacity (e.g., due to indoor living) struggle to maintain the Carnot-efficient temperature gradient, amplifying damage from blue light. Cold thermogenesis selectively benefits haplotypes with adaptable mitochondria (e.g., J or U), enhancing repair pathways, such as those involving Müller glia, while less adaptable haplotypes (e.g., H) remain vulnerable to a lack of repair. Cellular retinaldehyde-binding protein (CRALBP) supports production of 11-cis-retinaldehyde and its delivery to photoreceptors. It is found in the retinal pigment epithelium (RPE) and Müller glia (MG). Recent data reveal a dominant role for RPE-CRALBP in supporting rod and cone function, highlighting the importance of the RPE for cone regeneration. Unlike rods, cones have a unique requirement for rapid pigment regeneration to maintain their function in bright light and enable quick recovery after exposure to light.

- Carnot efficiency, derived from the Carnot cycle in thermodynamics, describes the maximum efficiency of a heat engine converting thermal energy into work, dependent on the temperature gradient between a hot source and a cold sink. In biological systems, mitochondria maintain a temperature gradient across membranes to optimize ATP production via oxidative phosphorylation (OxPhos). For Müller glia (retinal support cells) and the RPE (the pigmented layer that supports photoreceptors), a Carnot-efficient gradient enhances energy transduction, supports repair and regeneration, and maintains metabolic homeostasis. This gradient is disrupted by indoor living, which flattens thermal differences, atrophying mitochondrial colonies.
Why Eye Diseases Are More Common Today: Haplotypic and Thermodynamic Perspective
Blue Light Hazard and Mitochondrial Strain: Chronic ALAN liberates all-trans-retinal, generating ROS that tax mitochondrial defenses. Haplotypes with inefficient heat production (due to indoor warmth) and lower ROS scavenging (e.g., lighter melanin) are more susceptible to heat stress. The lack of UV/IR from sunlight, which boosts POMC and melanin regeneration, compounds this, as does nnEMF opening the blood-retinal barrier (Frey’s DARPA findings).
Carnot’s Efficiency Loss: Since indoor living flattens the temperature gradient, atrophying mitochondrial colonies in modern humans, haplotypes less adapted to this (e.g., those without cold-tolerance variants) experience greater energy deficits. This will impair retinal repair. Cold thermogenesis restores this gradient, but its efficacy varies by haplotype; individuals with thermogenic haplogroups may thrive, while others may lag behind. Heat production is tied to the Carnot Efficiency and directly alters the UPEs in the system (the thermodynamic limit of energy conversion). This varies by haplogroup and environmental context. This can change the noise in the system.
Thermogenic Capacity: Haplogroups like J, with cold-tolerance adaptations (e.g., UCP1 upregulation), maintain a temperature gradient via uncoupling, dissipating excess energy as heat. This supports Müller glia repair pathways under blue light stress, reducing ROS damage.
Indoor Living Impact: Modern indoor environments flatten this gradient, causing mitochondrial colonies to atrophy. Haplogroups without thermogenic variants (e.g., H) struggle to compensate, impairing energy supply to the retina. The Kowald and Kirkwood (2018) paper’s mtDNA deletion model suggests this stress could drive clonal expansion, worsening retinal decline.

- Cold Thermogenesis Benefit: Exposing mitochondria to cold restores the gradient, enhancing repair in adaptable haplogroups (e.g., J, U). Less adaptable haplogroups (e.g., H) show limited response, highlighting haplotype-specific resilience.
Blue light’s disruption of this gradient, by overexciting ETC and increasing ROS, further taxes non-thermogenic haplotypes, linking indoor living to retinal atrophy.
Regeneration Blockade: Sunlight’s UV/IR, absent indoors, drives Becker’s photo-bioelectric regeneration via DC currents and POMC. Haplotypes with robust melanin responses (e.g., L) regenerate better, while those with atrophied mitochondria (e.g., H under ALAN) face stalled repair, explaining rising disease prevalence and incidence.
Haplogroups influence melanin production via proopiomelanocortin (POMC) and melanocortin 1 receptor (MC1R) variants, affecting photoprotection:
Lighter Skin (e.g., Haplogroup H): Lower melanin in the RPE, driven by MC1R variants favoring lighter skin, reduces ROS scavenging and bioelectric conduction (Becker’s work). This heightens blue light damage, as melanin normally absorbs UV/visible light and emits ultraweak photon emissions (UPE) to signal repair.
Darker Skin (e.g., Haplogroup L): Higher melanin levels enhance photoprotection, neutralizing ROS and conducting bioelectric signals. However, in low-sunlight environments (e.g., northern U.S.), this advantage wanes, as melanin synthesis requires exposure to UV radiation. Migration disrupts this balance, explaining higher disease prevalence in African Americans as latitude rises despite innate resilience.
The decentralized thesis aligns here: melanin acts as an electromagnetic buffer, and its decline in mismatched environments (e.g., indoor technocracy) disrupts cellular coherence, increasing anterior chamber and retinal vulnerability.
- Why Eye Diseases Are More Common Today: The Technocratic Perspective
The increase in eye diseases, such as age-related macular degeneration (AMD), cataracts, and diabetic retinopathy, correlates with modern environmental and lifestyle shifts. Here’s a breakdown based on my research and related science:
- Shift to Artificial Light and Indoor Living:

- Most people now spend over 90% of their time indoors, exposed to artificial light (e.g., LEDs, screens) rather than sunlight. As our spectroscopes show, this light is dim and unbalanced compared to the sun’s full spectrum, which includes UV and infrared (IR) components essential for retinal health.
Artificial light at night (ALAN), rich in blue light (400-500 nm), disrupts the visual cycle. The chromophore all-trans-retinal, liberated from opsins during photoisomerization, accumulates when the retinal pigment epithelium (RPE) is overwhelmed, leading to oxidative stress and photoreceptor damage. This aligns with my “wrecking ball” metaphor for liberated vitamin A from the retina.
Blue light’s high energy penetrates the retina, exciting ETC complexes and generating ROS. Haplogroup-specific responses amplify disease risk:
Haplogroup H: Lower oxidative stress resilience and reduced melanin lead to increased ROS accumulation, heightening the risk of cataracts and age-related macular degeneration (AMD), especially indoors.
Haplogroup L0: Higher baseline ROS from ETC inefficiency, compounded by low sunlight in America, drives cataracts, diabetes (via mitochondrial dysfunction in pancreatic cells), and retinal diseases. The technocracy’s blue light dominance disrupts L0’s sunlight-dependent regeneration, explaining elevated rates in African Americans.
Haplogroup J: Thermogenic adaptability mitigates ROS, offering protection, but indoor living still poses a risk if sunlight exposure is insufficient.
The decentralized thesis suggests that blue light’s electromagnetic interference disrupts retinal coherence, a vulnerability that is magnified by haplotype-specific traits. Michael Rosbash’s 2017 Nobel Prize-winning insight, that the lack of sunlight during the day is worse than the artificial light at night, clearly supports my damaged retinal view, but he overlooks Becker’s work on regeneration. For me, the paper is important, but it misses a lot of quantum biology. Sunlight’s UV and IR are absent indoors, halting regeneration pathways that rely on POMC, melanin, and bioelectric signals (as per Becker’s work), thereby exacerbating damage.

- Evolutionary Context: Haplogroups evolved with environmental niches, H for Europe’s moderate climate, J for cold regions, L0 for high-sun tropics. Migration to mismatched environments (e.g., African Americans in the U.S.) unbalances these adaptations, increasing disease susceptibility as sunlight and melanin levels diverge from optimal.
Practical Solutions
Sunlight Exposure: 30–60 minutes of morning UV/infrared light boosts melanin synthesis and mitochondrial function, reducing ROS in all haplogroups. L0 benefits most with higher doses.
Blue Light Mitigation: Blue-blocking glasses or reduced screen time minimize ETC stress, especially for H and L0.
Cold Thermogenesis: Enhances mitochondrial gradient for J and U, supporting retinal repair, though less effective for H.
Melanin Support: Diets rich in tyrosine and copper (e.g., nuts, seafood) enhance POMC/MC1R activity, boosting photoprotection. When one integrates how melanin, iron, and blue light fit into this decentralized thesis, you have to leverage insights from this paper in the Biomedical Journal of Scientific & Technical Research
The paper (https://biomedres.us/pdfs/BJSTR.MS.ID.008142.pdf) explores their roles in evolutionary adaptation, electromagnetic coherence, and disease. The paper reframes melanin’s evolutionary role from photoprotection to a metal-chelating agent, particularly for iron, facilitating the excretion of heavy metals via epidermal turnover in humans. This aligns with my decentralized thesis by positioning melanin as a dynamic electromagnetic black box regulator in eukaryotes.
Melanin was an Evolutionary Driver: Dietary shifts in early humans introduced excess iron, driving melanin synthesis in melanocytes to bind and excrete it through desquamation. This explains racial pigmentation differences, with darker skin (phototypes IV-VI) evolving in high-iron environments, which supports the view that melanin acts as a calibrator of environmental inputs.
Iron Homeostasis: Melanin-bound iron loss through skin turnover depletes systemic iron, increasing the risk of anemia in heavily melanated individuals, as noted in the paper. This challenges the centralized “sunscreen” model, framing melanin as a redox and electromagnetic stabilizer in humans
Quantum Interface: Melanin’s lattice absorbs all light frequencies and chelates iron without reflection, converting electromagnetic energy into bioelectric signals, consistent with my black body analogy.
The thesis highlights blue light’s role in shifting iron to Fe²⁺, generating nitric oxide (NO) and localized hypoxia, while the paper adds melanin’s amplifying quantum effect:
Photoexcitation: Blue light (380–450 nm) excites melanin, triggering a one-electron transfer that produces ROS (superoxide, hydrogen peroxide). Iron-saturated eumelanin enhances this, broadening near-infrared absorption and intensifying oxidative stress, disrupting cellular redox fields. This alters UPE signaling.
Retinal Impact: In the RPE, melanin binds iron to mitigate ROS, but blue light’s interaction with this complex increases UPE and oxidative damage. This aligns with my mitoception thesis, where UPE signals mitochondrial stress (e.g., via GDF15), contributing to eye diseases.
ECS Disruption: Blue light’s oxidative assault stresses the ECS (Di Meo et al., 2025), impairing electromagnetic stability and amplifying mitochondrial dysfunction, a key theme in this framework. This would also alter UPEs in the ECS system.
Blood-Retinal Barrier Disruption by nnEMF:
Allan Frey’s 1960s DARPA research demonstrated that non-native electromagnetic fields (nnEMF) from technologies (e.g., microwaves, Wi-Fi) can open the blood-retinal barrier by affecting endothelial tight junctions. This increased permeability allows toxins and inflammatory molecules to reach the retina, heightening susceptibility to damage from ALAN and free retinal.
This mechanism is less emphasized in centralized ophthalmology, which focuses on genetic or aging factors; however, it supports my claim of environmental exacerbation due to changes in the ionosphere’s conductance, which are radically inducing eye pathology. The establishment’s centralized narratives clearly downplay nnEMF’s role due to industry interests, a point worth questioning because their incentives are driving chronic eye disease epidemics.

- Evolutionary Context: Haplogroups evolved with environmental niches, H for Europe’s moderate climate, J for cold regions, L0 for high-sun tropics. Migration to mismatched environments (e.g., African Americans in the U.S.) unbalances these adaptations, increasing disease susceptibility as sunlight and melanin levels diverge from optimal.
Practical Solutions
Sunlight Exposure: 30–60 minutes of morning UV/infrared light boosts melanin synthesis and mitochondrial function, reducing ROS in all haplogroups. L0 benefits most with higher doses.
Blue Light Mitigation: Blue-blocking glasses or reduced screen time minimize ETC stress, especially for H and L0.
Cold Thermogenesis: Enhances mitochondrial gradient for J and U, supporting retinal repair, though less effective for H.
Melanin Support: Diets rich in tyrosine and copper (e.g., nuts, seafood) enhance POMC/MC1R activity, boosting photoprotection. When one integrates how melanin, iron, and blue light fit into this decentralized thesis, you have to leverage insights from this paper in the Biomedical Journal of Scientific & Technical Research
The paper (https://biomedres.us/pdfs/BJSTR.MS.ID.008142.pdf) explores their roles in evolutionary adaptation, electromagnetic coherence, and disease. The paper reframes melanin’s evolutionary role from photoprotection to a metal-chelating agent, particularly for iron, facilitating the excretion of heavy metals via epidermal turnover in humans. This aligns with my decentralized thesis by positioning melanin as a dynamic electromagnetic black box regulator in eukaryotes.
Melanin was an Evolutionary Driver: Dietary shifts in early humans introduced excess iron, driving melanin synthesis in melanocytes to bind and excrete it through desquamation. This explains racial pigmentation differences, with darker skin (phototypes IV-VI) evolving in high-iron environments, which supports the view that melanin acts as a calibrator of environmental inputs.
Iron Homeostasis: Melanin-bound iron loss through skin turnover depletes systemic iron, increasing the risk of anemia in heavily melanated individuals, as noted in the paper. This challenges the centralized “sunscreen” model, framing melanin as a redox and electromagnetic stabilizer in humans
Quantum Interface: Melanin’s lattice absorbs all light frequencies and chelates iron without reflection, converting electromagnetic energy into bioelectric signals, consistent with my black body analogy.
The thesis highlights blue light’s role in shifting iron to Fe²⁺, generating nitric oxide (NO) and localized hypoxia, while the paper adds melanin’s amplifying quantum effect:
Photoexcitation: Blue light (380–450 nm) excites melanin, triggering a one-electron transfer that produces ROS (superoxide, hydrogen peroxide). Iron-saturated eumelanin enhances this, broadening near-infrared absorption and intensifying oxidative stress, disrupting cellular redox fields. This alters UPE signaling.
Retinal Impact: In the RPE, melanin binds iron to mitigate ROS, but blue light’s interaction with this complex increases UPE and oxidative damage. This aligns with my mitoception thesis, where UPE signals mitochondrial stress (e.g., via GDF15), contributing to eye diseases.
ECS Disruption: Blue light’s oxidative assault stresses the ECS (Di Meo et al., 2025), impairing electromagnetic stability and amplifying mitochondrial dysfunction, a key theme in this framework. This would also alter UPEs in the ECS system.
Blood-Retinal Barrier Disruption by nnEMF:
Allan Frey’s 1960s DARPA research demonstrated that non-native electromagnetic fields (nnEMF) from technologies (e.g., microwaves, Wi-Fi) can open the blood-retinal barrier by affecting endothelial tight junctions. This increased permeability allows toxins and inflammatory molecules to reach the retina, heightening susceptibility to damage from ALAN and free retinal.
This mechanism is less emphasized in centralized ophthalmology, which focuses on genetic or aging factors; however, it supports my claim of environmental exacerbation due to changes in the ionosphere’s conductance, which are radically inducing eye pathology. The establishment’s centralized narratives clearly downplay nnEMF’s role due to industry interests, a point worth questioning because their incentives are driving chronic eye disease epidemics.
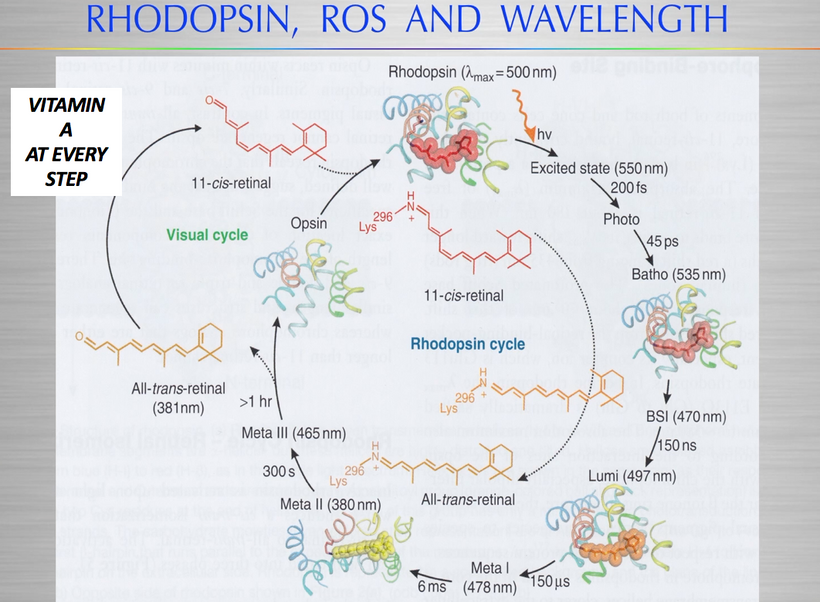
- Integration of these ideas into the Decentralized Thesis
Light as Primacy: Melanin’s light absorption and iron chelation convert environmental photons into bioelectric currents, regulating mitochondrial coherence. Blue light, however, disrupts this, shifting iron oxidation states while generating ROS, which my thesis identifies as a root cause of systemic imbalance.
Electromagnetic Coherence: Melanin and iron form a quantum interface, modulating EMF interactions. Blue light’s interference with this system, via ROS and UPE, destabilizes cellular fields, necessitating ECS-mediated repair.
Evolutionary Context: Following the Post-GOE period, melanin’s iron-binding capacity evolved to manage oxidative stress resulting from increased oxygen levels, integrating with mitoception and the leptin-melanocortin pathway.
Regeneration Blockade:
The visual cycle, which regenerates 11-cis-retinal from all-trans-retinal, depends on sunlight’s UV and IR to stimulate POMC and melanin production. POMC-derived peptides (e.g., α-MSH) enhance melanin’s photoprotective and regenerative roles, while Becker’s bioelectricity research suggests UV/IR-induced DC currents are needed to activate retinal repair (e.g., Müller glia = eye stem cells need NO). Glaucoma is also related to these effects of a lack of UV-IR on eye pressure gradients.

- Indoor living blocks these pathways, as you argue, preventing regeneration. The atrophy of mitochondria, our “hydrogen heat engines”, due to lack of seasonal temperature variation (e.g., cold thermogenesis) further impairs energy for repair, aligning with Carnot’s efficiency principle: greater temperature gradients enhance engine performance.
Tech Addiction and Neurochemical Disruption
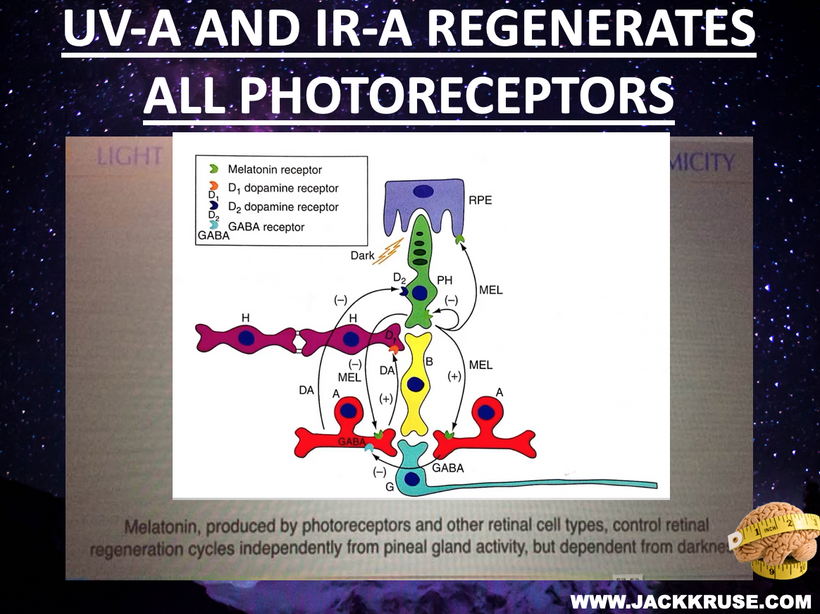
My “zombie” analogy reflects how screen addiction, driven by blue light’s temporary dopamine boost, disrupts melatonin and GABA signaling (slide below). This circadian misalignment, compounded by indoor confinement, reduces retinal health and systemic resilience.
Casinos and tech companies exploit this, designing blue-rich environments to sustain engagement, given dopamine’s role in the brain as a reward.
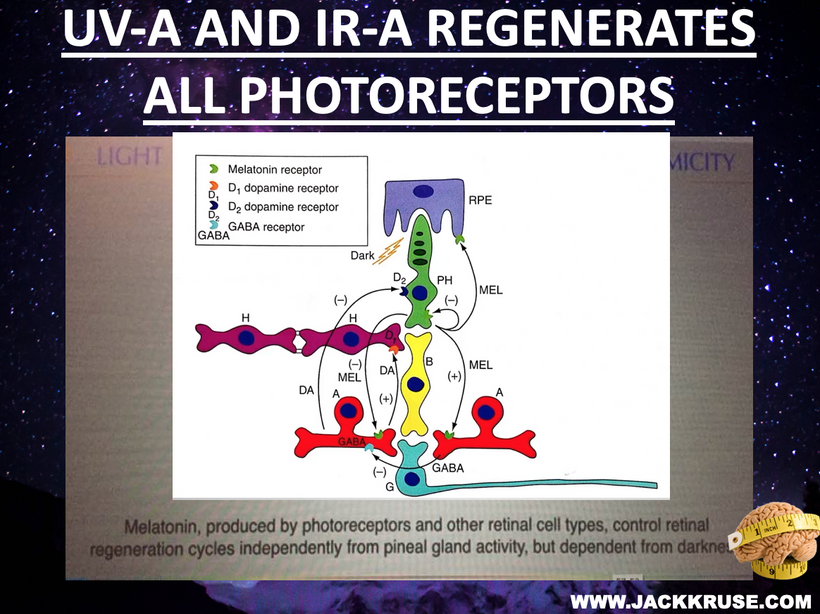
- Mitochondrial Failure and Thermodynamic Loss
Mitochondria in retinal cells, with high oxygen demand, are vulnerable to blue light-induced ROS, as noted in recent studies on AMD. My Carnot efficiency argument suggests that hotter engines (mitochondria) run more efficiently, which implies that cold thermogenesis boosts mitochondrial function by increasing the temperature gradient, thereby countering atrophy from thermostat-controlled indoor life.
This thermodynamic perspective is unconventional in medicine but aligns with Becker’s bioelectric findings and my call to reconnect with nature’s frequencies.

The Centralized narrative often attributes rising eye disease prevalence to aging, genetics, and lifestyle factors like diabetes, overlooking environmental drivers like ALAN, nnEMF, and sunlight deficiency. The destruction of heme, along with all our other photoreceptors, explains fully why modern chronic disease epidemics are present.
The synergy of melanin, iron, and blue light defies reductive biochemical models, embodying a Camus-esque revolt. This thesis empowers individuals to reclaim coherence by aligning with natural light and EMF, countering the technocracy’s disruptive influence. Melanin’s iron-chelating role is amplified by the oxidative effects of blue light, allowing for a perfect integration into this decentralized thesis as a quantum interface regulating electromagnetic coherence. Evolving post-GOE, it supports mitoception via UPE and GDF15, with blue light disrupting this balance in diseases such as AMD and mental health disorders. Restoring natural light and minimizing disruptors restores this system, aligning with my vision of biological agency.
HOW DID THIS HAPPEN?
F1-ATPase explains how this all happened. Here are the basics explaining how uncoupled haplotypes evolved due to variable EMFs as latitude and longitude changed: F1-ATPase is a rotary molecular motor within mitochondria that synthesizes ATP by harnessing the proton motive force across the mitochondrial membrane. Protons flow through the enzyme’s F0 subunit, driving the F1 subunit’s rotation (the “spin rate”) to catalyze ATP production from ADP and inorganic phosphate.

The cited paper in Figure 1 above models F1-ATPase’s dynamics, showing it’s highly reversible: it can synthesize ATP when rotating forward (hydrolysis-driven) or hydrolyze ATP when forced backward. However, under high external torque, caused by stressors such as oxidative damage due to excessive oxygen, insufficient sunlight, or a lack of melanin resulting from environmental changes, a metabolic imbalance occurs. This enzyme undergoes “mechanical slip,” where rotation occurs without effective ATP production, resulting in the dissipation of energy as heat. This is precisely how uncoupled haplotypes were innovated as humans left their tropical habitats to conquer colder climates.
The “mechanical slip” is a state in which the enzyme rotates without effectively producing ATP, instead dissipating energy as heat. This inefficiency, noted in the paper as reducing free energy transduction by 40–80% below optimal, reflects a breakdown in chemomechanical coupling under nnEMF light stress. Light stress leads to an increase in deuterium concentration, causing a mechanical shift in the ATPase. LIGHT > FOOD
Environmental Stressors and Torque on F1-ATPase
As humans migrated out of the tropics (~200,000 years ago), they encountered varying EMFs, latitudes, and longitudes, altering sunlight exposure, oxygen levels, and melanin production. These changes acted as external torques on F1-ATPase:
Sunlight Reduction: Tropical regions receive abundant UV and infrared light, which optimizes mitochondrial function through photobiomodulation (e.g., enhancing cytochrome c oxidase activity). At higher latitudes, reduced sunlight, especially UV, disrupted this, lowering ATP efficiency and increasing ROS, a stressor that torques F1-ATPase.
Oxygen Variability: Colder climates often have higher oxygen partial pressures, which enhances ROS production during OxPhos. Excessive oxygen, as a stressor, should force F1-ATPase into reverse or slip modes, dissipating energy.
Melanin Decline: In the tropics, high melanin levels protected against UV and stabilized redox balance, supporting F1-ATPase efficiency. As humans moved to northern latitudes with less UV, melanin production decreased (e.g., lighter skin evolved), reducing antioxidant capacity and exacerbating oxidative stress, further torquing the enzyme to slip more.
Sleep Mechanics and eye diseases: GDF15 signals mitochondrial stress (mitoception), which causes a rise with electron surplus, prompting sleep as a mitoceptive response. Melanin’s UPE release, shared with GDF15, reinforces this photonic feedback loop in humans. My thesis posits that cellular health depends on “field stability” in tissues. An electron surplus disrupts this, and sleep is driven by UPEs and melanin’s photonic feedback, restoring it. Melanin’s UPE emission, tied to ROS and iron, signals this surplus, linking this signal to mitoception. The GOE enabled OxPhos and UPEs to manifest, thereby driving the electromagnetic origins of sleep. Leptin’s 220 nm absorption, tied to endogenous light transformation, evolved to regulate this surplus, integrating with the leptin-melanocortin pathway. Red light’s role in mito-dR supports my light-centric view, countering the disruptive effects of blue light (e.g., ROS from melanin-iron complexes). This aligns with the retina’s glycolysis adaptation under photonic stress. (seen below)

This environmental shift created a metabolic imbalance, where the spin rate of F1-ATPase struggled to match energy demands, leading to mechanical slip and heat dissipation rather than ATP synthesis.
The innovation of uncoupled haplotypes involved mtDNA mutations affecting F1-ATPase or UCP expression:
Mutation Selection: Random mtDNA deletions or point mutations, as modeled in the cited paper, should disrupt F1-ATPase’s coupling efficiency, favoring slip under torque. Over generations, these variants were selected in cold climates where heat generation was prioritized over ATP maximization.
UCP Upregulation: UCPs (e.g., UCP1 in brown fat) evolved to uncouple OxPhos, dissipating PMF as heat. The torque-induced slip phenotype prefigured this out, with natural selection amplifying UCP expression in uncoupled haplotypes. Natural selection rendered melatonin’s function less important for overall energy balance and expanded the leptin-melanocortin pathway because it was more accurate. Today, melatonin production in the mtDNA is the only remnant of its GOE importance.

Geographic Divergence: Haplogroups (e.g., H in Europe, M in Asia) reflect these adaptations, with northern populations exhibiting higher uncoupling efficiency (e.g., via UCP1 polymorphisms) to counteract low sunlight and cold temperatures. The paradox of haplotype vulnerability, a strength in one environment that becomes a weakness in another, mirrors a dead man walking who seeks Bitcoin. My decentralized approach rebels against the technocracy’s indoor, blue-light-dominated paradigm, advocating light and coherence to reclaim retinal health. This aligns with Sartre’s concept of freedom: individuals can choose to align their environment to mitigate haplotype-specific risks.

SUMMARY
Mitochondrial haplogroups (H, J, L0) influence metabolic efficiency, melanin production, and photoreceptor health, modulating blue light vulnerability. H and L0 face higher risks due to oxidative stress and melanin mismatch in indoor settings, while J’s thermogenic resilience offers protection.
The technocracy’s disruption of sunlight and temperature gradients exacerbates this, driving diseases like cataracts and AMD. Restoring natural light and thermogenesis can mitigate these effects, empowering haplotype-specific resilience.
I challenge this narrative by linking these to industrial and technological agendas (e.g., DARPA’s nnEMF research, tech addiction), suggesting a deliberate blinding and regeneration suppression. While evidence supports ALAN’s role in AMD and nnEMF’s barrier effects, the regeneration claims (e.g., UV/IR, Becker’s work) are less mainstream, though emerging research on melanin’s protective role and optogenetic repair lends credence to explaining the modern disease epidemics linked to the eye.
Eye diseases are more common today due to chronic exposure to ALAN and nnEMF, which damage the retina and open the blood-retinal barrier, combined with the loss of sunlight’s UV and IR, which halt regeneration via POMC, melanin, and bioelectric pathways. Mitochondrial atrophy from indoor life further compounds this, as per Carnot’s efficiency principle. Reconnecting with nature, through sunlight and cold thermogenesis, will restore these processes, countering the degenerative effects of artificial frequencies and tech addiction.
Sleep in humans is triggered by an electron surplus, creating charge density, and acts as a cellular sweep to restore electromagnetic coherence, which is signaled by adenosine and modulated by UCP4, red light, and sesB. This aligns with my decentralized thesis, connecting to melanin’s UPE, mitoception’s GDF15, and the primacy of light in most diseases.
CITES
https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2024.1509434/full
https://www.linkedin.com/pulse/blinding-us-while-stopping-our-regeneration-jack-kruse-6jsze