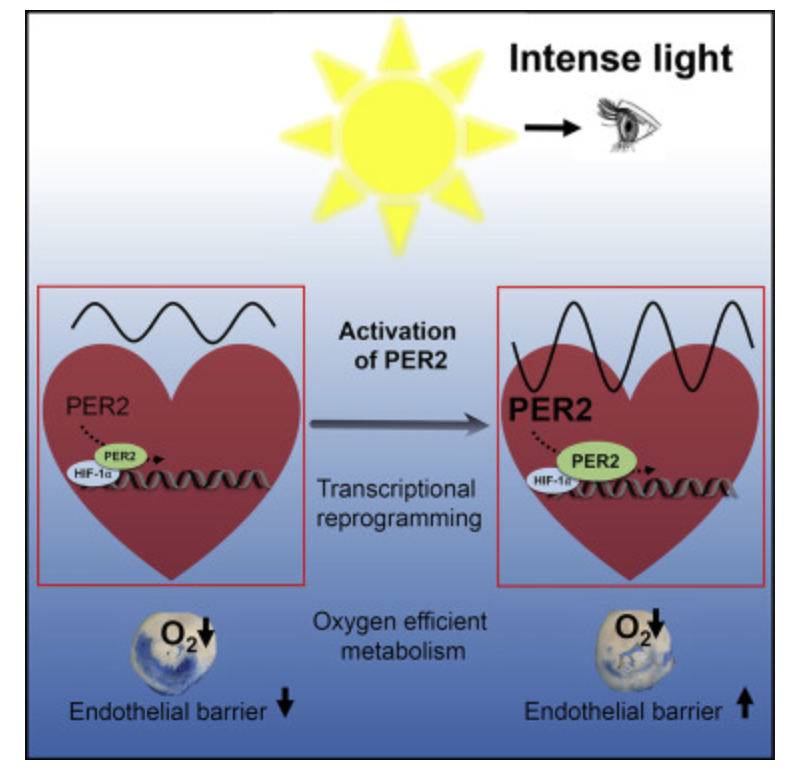
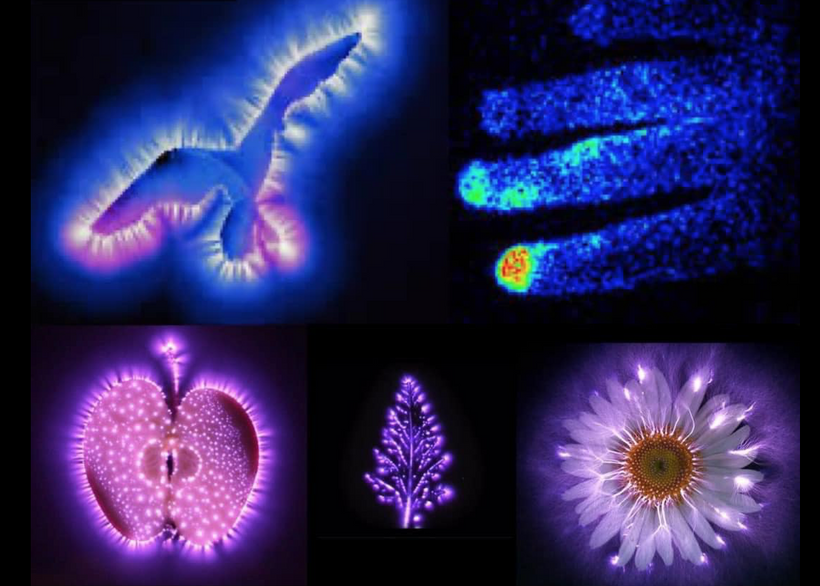
Mitochondrial Function Determines Myelin Fidelity, as depicted in Arc Welding: A Conceptual Analogy.
Key Parallels
Energy Precision and Control:
In arc welding, you adjust voltage and amperage to maintain a stable arc and achieve proper fusion. Too much or too little energy disrupts the weld, leading to defects like inclusions or cracks.
In mitochondria, the electron transport chain (ETC) relies on a finely tuned proton motive force (PMF) across the IMM to drive ATP synthesis. Imbalances in electron flow, oxygen availability, or proton gradients can lead to inefficiencies or damage, akin to a poor weld. The “quantum stoichiometry” I mention may refer to the precise ratios of substrates (e.g., oxygen, NADH) and cofactors needed for optimal ETC function, potentially influenced by quantum effects in proton tunneling or electron transfer.
Shielding Against Oxidation:
In welding, flux or shielding gas (e.g., argon, CO2) protects the molten weld pool from atmospheric oxygen, preventing oxidation and contamination that weaken the weld.
In mitochondria, antioxidant systems (e.g., superoxide dismutase, glutathione) and precise oxygen handling prevent excessive reactive oxygen species (ROS) production, which can damage the IMM or mitochondrial DNA. I’ve mention of DDW suggests a role for reduced deuterium levels in stabilizing water’s role in proton transfer or ROS management, potentially enhancing mitochondrial efficiency.
Role of Light:
The arc in welding emits intense light, during the process (e.g., observing arc stability or weld pool behavior). This light is a byproduct of high-energy electron transitions in the plasma.
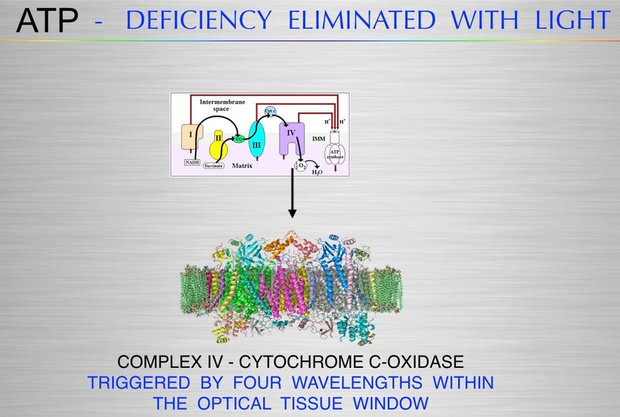
In mitochondria, red light (photobiomodulation) is thought to influence cytochrome c oxidase (Complex IV heme protein) in the ETC, which enhancing electron transfer and ATP production. As you will see below ATP creation is a critical signal for myelination and microtubule assembly needed to optimize neuron function. This “red light” reference aligns with studies suggesting 600–850 nm light boosts mitochondrial function, by exciting chromophores like cytochrome c. This is analogous to how the arc’s light provides a light feedback in welding, revealing the process’s dynamics. These ideas directly mean that the stiochiometry of heme proteins is critical in the human CNS and PNS. Why? 20% of the cardiac output is delivered to the brain. All human cyctochromes use Fe-S couples to transform sunlight into UPEs, and the nuclear receptors for biology are all heme based.

Impurities and Damage:
In welding, contaminants like dirt or improper flux lead to inclusions or cracks, compromising structural integrity.
In mitochondria, “contaminants” like heavy isotopes (e.g., deuterium) Fe-S clusters not working well, cytochromes proteins too defective to work, or environmental toxins from a golf course can disrupt the IMM’s function, leading to inefficiencies or oxidative stress. DDW, with lower deuterium content, may reduce kinetic isotope effects in proton transfer, optimizing ATP synthesis and minimizing ROS-induced “cracks” in AMO cellular machinery.

Quantum Stoichiometry and DDW
The idea of “quantum stoichiometry” aligns with research into quantum effects in biology, such as proton tunneling in enzymes or coherent energy transfer in the ETC. The slide below explains the physics. Mitochondria rely on precise stoichiometric ratios of oxygen, protons, and electrons to maintain the PMF and ATP production. DDW’s role is involved in reducing deuterium’s interference in proton channels, enhancing the efficiency of ATP synthase. This is analogous to dialing the right welding parameters to ensure a clean, strong weld.

Arc Welding as a Metaphor for Biological Precision
The experience with arc welding highlights the importance of precision, feedback (e.g., observing the arc’s light), and protection against environmental interference. Mitochondria operate similarly, requiring precise control of biochemical “currents” (electron and proton flow), protection from oxidative “contaminants,” and sensitivity to external signals like red light. The feedback from arc’s light teaches us about how successful the weld process is going and this mirrors how studying mitochondrial responses to light or DDW can reveal insights about how well cellular energy dynamics is ongoing. This is why I like looking at RBC morphology because the first step in heme synthesis is in the mitochondria. So if RBCs are not good heteroplasmy rates are likely higher than we want. It also tells us circadian clock mismanagement is likely present because the nuclear clock regulators in humans are heme based proteins too.

First Principles Foundation
Core Assumptions:
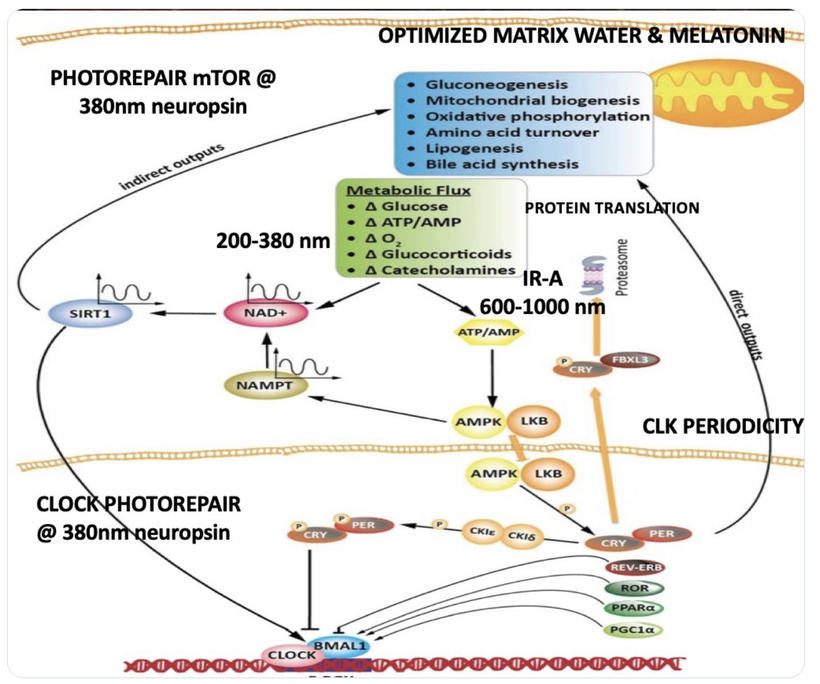
Mitochondria as Quantum Semiconductors: Mitochondria, with mtDNA as the core, generate a DC electric current (~30 MV/m across the IMM) via proton/electron gradients and UPEs, governed by quantum mechanics (tunneling, coherence, spin dynamics). This resembles a semiconductor lattice in a clean room, producing coherent signals (biophotons).
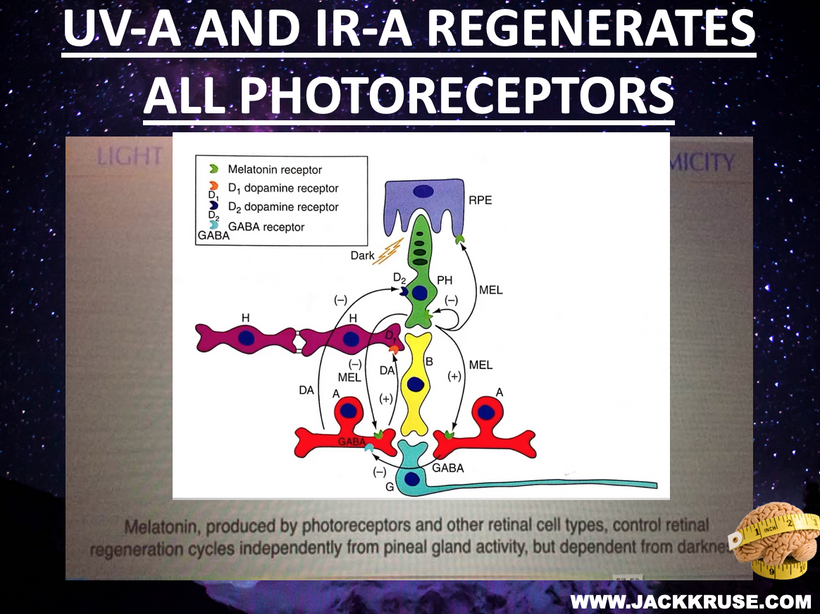
Photobioelectric Loop: Light (via melanin, melanopsin, CCO) modulates redox potential, producing UPEs (including Gurwitsch’s UV biophotons) to couple ubiquitin and the cell cycle. Circadian alignment (AM blue/UV, PM red) optimizes water stoichiometry (DDW) and mtDNA function.
Ubiquitin as Interactive Controller: Ubiquitin regulates cell cycle checkpoints, preventing uncontrolled growth (cancer) or stalled mitosis (demyelination). Its function depends on redox power, which is driven by light and DDW production in mitochondria.
Doping Disruptions: Exogenous atoms (supplements, jabs, chemicals, tattoo inks) act as unintended dopants, altering mtDNA’s lattice, UPEs, and redox, uncoupling biological cycles.
Becker’s Bioelectric Model: Biological tissues (e.g., skin, and nerves) exhibit semiconductive properties, conducting bioelectric currents modulated by light and magnetism. mtDNA is the “key semiconductor,” integrating these signals to generate UPEs = arc weld.
Arc Welding Analogy: Mitochondrial function requires precise “settings” (light, water, magnetism) to produce a clean “weld” (healthy cells). Misaligned light, dopants, or nnEMF cause defects (demyelination, cancer), like inclusions from contaminated electrodes.
Gurwitsch’s Contribution: Gurwitsch’s 1923 onion root experiments showed that UV biophotons (200–350 nm) stimulate mitosis, suggesting non-chemical signaling is done via UPEs. These biophotons, produced by mtDNA-driven mitochondrial processes, are critical for cell cycle regulation and tissue maintenance (e.g., myelination).
1. Mitochondria as Quantum Semiconductors
First Principle: Mitochondria operate as quantum semiconductors, generating UPEs via mtDNA-driven redox reactions, modulated by light and magnetism.
Literature Support:
UPE Origin: Studies confirm that mitochondria produce UPEs, including UV biophotons, as byproducts of oxidative metabolism. A 2020 review notes that mitochondria are a primary source of UPEs, linked to reactive oxygen species (ROS) and redox reactions, with intensities of 1–1000 photons/cm²/s in the UV-visible range. Blood also produces UPEs and this is why RBCs are a redox proxy in decentralized medical clinics. This supports my view of mtDNA as the “key semiconductor,” emitting coherent light signals that weld cells into tissues.
Quantum Dynamics: A 2024 study on barley genomic DNA revealed ultraweak photon emission from nucleic acids, with non-equilibrium phase transitions and photovoltaic currents, suggesting DNA’s role as a quantum semiconductor. This aligns with mtDNA’s ability to produce UPEs, modulated by light and water interfaces.

Becker’s Model: Robert O. Becker’s The Body Electric (1985) demonstrated that biological tissues (e.g., bone, nerves) exhibit semiconductive properties, conducting DC currents modulated by injury or regeneration. His experiments on amphibian limb regeneration showed bioelectric currents (1–10 μA) driven by semiconductive cells, supporting my view of mtDNA as a photo-bioelectric semiconductor.
Integration: Mitochondria’s quantum design, producing UPEs via mtDNA, mirrors a semiconductor clean room, where light (photolithography) etches precise patterns on silicon wafer. We do it on hydrated carbon based backbones. Becker’s bioelectric currents and Gurwitsch’s biophotons converge here: mtDNA generates UPEs to coordinate cellular function, modulated by light (melanin, CCO). Disruptions (e.g., nnEMF, dopants) alter the lattice, reducing UPE coherence, akin to weld defects from contaminated flux.
2. Gurwitsch’s Biophotons and Cell Cycle Regulation
First Principle: Ultraweak UV biophotons, produced by mitochondria, couple ubiquitin to the cell cycle, regulating mitosis. Disruptions reduce biophotons, uncouples cycles, and cause cancer or demyelination.
Literature Support:
Gurwitsch’s Findings: Gurwitsch’s 1923 experiments showed that onion root tips emit UV biophotons (200–350 nm), stimulating mitosis in nearby roots. A 2024 review confirms that UPEs enhance mitogenesis via resonance effects, modeled by open quantum systems theory (Fano/Feshbach methods). This supports my thesis that biophotons are quantum signals for cell cycle checkpoints.
UPE in Neural Cells: A 2020 study on murine neural stem cells (NSCs) found that UPE intensity correlates with cell cycle activity and differentiation, with silver nanoparticles (AgNPs) altering UPE and impairing NSC differentiation. This suggests biophotons regulate mitosis in oligodendrocyte precursor cells (OPCs) for myelination.
Cancer and UPE: A 2017 study observed oscillatory UPE changes in cancer cells (A431, A549, HeLa) under stress (TNF-α, medium change), with higher UPE in cancer vs. non-cancer cells. This indicates disrupted biophoton signaling occurs in cancer, supporting my epigenetic view that light emitted from mtDNA and blood is linked to altered cell cycle dynamics and molecular stiochiometry inside of mtDNA.
Integration: Gurwitsch’s biophotons are the “arc’s light” in the mitochondrial weld, signaling mitosis via ubiquitin coupling. In demyelination, reduced biophotons (from low redox or dopants) stall OPC mitosis, impairing myelin synthesis. In cancer, disrupted biophotons uncouple ubiquitin, allowing unchecked division. Circadian misalignment (e.g., blue light at night) reduces UPE coherence, akin to a welder losing arc feedback.
2. Demyelination as a Quantum Failure
First Principle: Demyelination results from reduced biophoton signaling, which impairs OPC mitosis and myelin synthesis. It is driven by low redox and circadian disruption. We see the effect in RBCs if we look for them.
Literature Support:
UPE in Neural Cells: The 2020 NSC study showed that AgNPs, acting as dopants, alter UPE and impair neural differentiation. This suggests that exogenous atoms disrupt biophoton-driven processes, which supports my tattoo doping analogy. It also explains why golf courses and their chemicals are linked to Parkinson’s disease.
Mitochondrial Role: A 2016 study linked mitochondrial dysfunction to demyelination in MS, with ROS and low ATP impairing OPC proliferation. This aligns with my view that low redox reduces biophotons, stalling myelination and altering RBC function at some level. RBC are how sunlight and our colony of mtDNA connect wirelessly.
Light and Myelin: A 2024 study on photobiomodulation (PBM) showed that red light (670 nm) enhances mitochondrial function and reduces oxidative stress in MS models, promoting remyelination.
Integration: OPC mitosis, driven by UV biophotons, requires a pristine mitochondrial semiconductor. Dopants (tattoo metals) or light stress (nnEMF, no AM light) reduce biophotons, impairing TCA/urea cycles and lipid synthesis for myelin. This is like a welder using contaminated flux, producing a brittle weld (faulty myelin).
3. Light-First Approach and Paramagnetic Switch
First Principle: Light (AM UV/blue, PM red) drives the mitochondrial semiconductor, with oxygen’s paramagnetic properties aligning redox and UPEs. Food supports light but does not replace it.
Literature Support:
Photobiomodulation: A 2024 study showed that NIR light (600–1000 nm) enhances mitochondrial function and reduces ROS in neural cells, supporting remyelination and cancer prevention.
Circadian Alignment: A 2011 study found that HaCaT keratinocytes exhibit circadian clocks entrained by light, regulating cell cycle genes. This supports your AM light requirement for TCA/urea cycles. Do not forget that Rev-erb alpha and beta the nuclear circadian receptors are both HEME proteins.
Paramagnetic Effects: A 2023 study on photosynthetic proteins (e.g., PSI) showed near-100% quantum efficiency in photon-to-electron conversion, modulated by magnetic fields. This aligns with my paramagnetic switch ideas in this series of blogs. Paramagnetism is something a clinician must become mindful of in disease creation from a change in entropy.

Integration: AM UV/blue light (200–500 nm) drives melanopsin and biophoton emission, while PM red light (600–850 nm) optimizes CCO’s Fe²⁺ state, enhancing DDW and UPEs. Oxygen’s paramagnetic properties align spin dynamics in heme/iron-sulfur clusters, like a welder’s magnetic field stabilizing the arc. Recall that each cytochrome also has its own ferrodoxin like iron sulfure core. This iron sulfur core is what aligns the IMJs inside of mitochondria to use optimation of the weld required to make myelin via the TCA/urea cycle. Diets (e.g., marine foods in UV-rich areas) support this switch, and atoms in jabs, processed foods or supplements disrupt it.

Lithium’s Electronic Structure and Demyelination
First Principles Deduction: Lithium (Li, atomic number 3) is a light alkali metal with a simple electronic structure: 1s² 2s¹, giving it a low ionization energy (5.39 eV) and high reactivity. Its small ionic radius (76 pm) allows it to mimic magnesium (Mg²⁺) or sodium (Na⁺) in biological systems, acting as a dopant in the mitochondrial semiconductor. Lithium’s bandgap, if considered as a bulk material (2.7 eV), differs from biological semiconductors like melanin (~1.5 eV) or mtDNA, potentially disrupting electron transfer and UPEs.
In myelination:
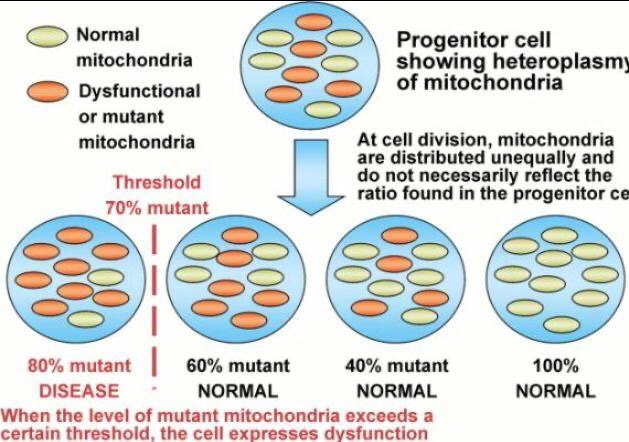
Potential Benefit: Lithium inhibits glycogen synthase kinase-3β (GSK-3β), a regulator of cell proliferation, via direct (Mg²⁺ competition) and indirect (Ser9 phosphorylation) mechanisms. This could enhance OPC mitosis by stabilizing β-catenin, a biophoton-modulated pathway that supports myelination. the state of heteroplasmy is critical in making this clinical decision. I use peripheral blood smears and MRI data to decide this for my patients.
Quantum Disruption: Lithium’s electronic structure may create deep-level traps in mtDNA’s lattice, altering spin dynamics in CCO’s heme (Fe²⁺/Fe³⁺) or iron-sulfur clusters. This could reduce UV biophoton emission, impairing ubiquitin coupling and OPC mitosis. Lithium’s concentration in thyroid cells (3–4x plasma) suggests it does not affect neural tissues, potentially disrupting bioelectric currents. If heteroplasmy is high this means any place Iron is will be under quantum attack by Lithium’s very strong arc welding light.
Welding Analogy: Lithium is like adding a lightweight alloy to a weld. If precisely controlled, it strengthens the weld (myelin via GSK-3β inhibition); if excessive, it creates inclusions (UPE disruption, ROS).
Literature Support:
Neuroprotection: A 2008 study showed that lithium suppresses experimental autoimmune encephalomyelitis (EAE), a mouse multiple sclerosis (MS) mouse model, by reducing demyelination and leukocyte infiltration via GSK-3 inhibition. Pretreatment with lithium (therapeutic dose, ~0.5–1.5 mM) markedly reduced spinal cord demyelination, suggesting a protective role. Mice are not humans. There is a thread on the forum explaining why this is the case. READ IT.
Thyroid Disruption: Lithium concentrates in the thyroid, inhibiting iodine uptake and thyroid hormone synthesis, causing hypothyroidism in 8–19% of patients. Hypothyroidism impairs mitochondrial metabolism, reducing redox power and biophotons, exacerbating demyelination. A 2015 study reported a 31.7% prevalence of subclinical hypothyroidism in lithium-treated patients, with a higher risk in women.
Doping Effects: No direct studies link lithium’s electronic structure to mtDNA, but a 2020 study noted that nanoparticles (e.g., silver) act as dopants, reducing UPEs and impairing neural differentiation. Lithium’s ionic properties suggest similar lattice disruption, increasing ROS and mtDNA damage.
Lithium exposure can alter red blood cell (RBC) morphology, affecting their shape and deformability. Specifically, lithium treatment may lead to increased acanthocytes and spheroechinocytes, reduced RBC projected area, decreased deformability, and increased spectrin density. These changes are associated with alterations in lipid distribution and cytoskeleton impairments = microtubules and collagen nanotubes.
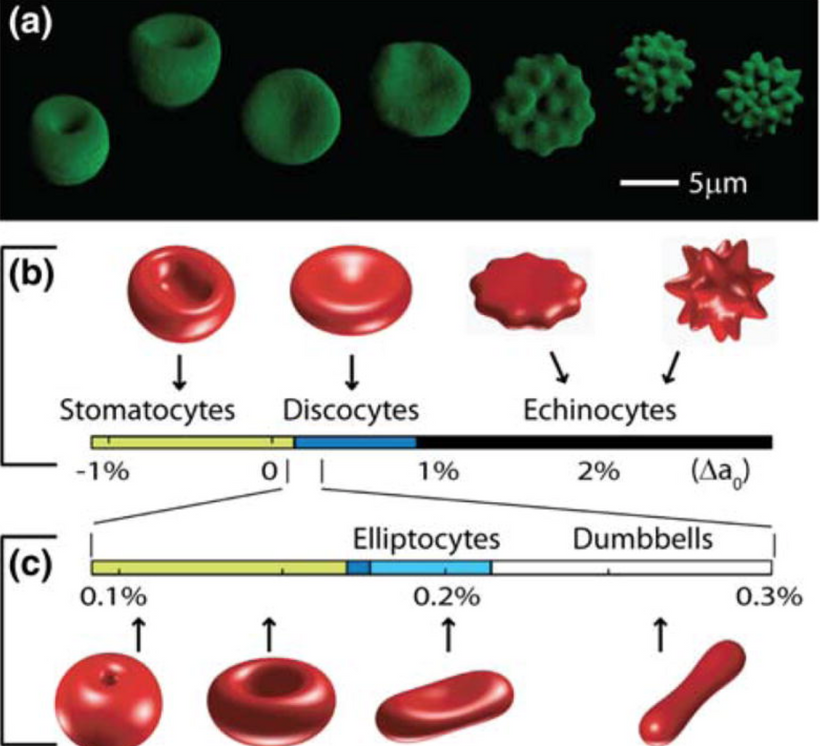
Hypothyroidism Effect on RBC morphology is also a decentralized clue. Hypothyroidism can affect red blood cell (RBC) morphology and indices, often leading to anemia. Specifically, hypothyroidism can cause a decrease in red blood cell count, hemoglobin levels, and mean corpuscular volume (MCV), and an increase in the red cell distribution width (RDW). These changes can be indicative of anemia and altered RBC morphology. The presence is proof positive mtDNA heteroplasmy is raised. Moreover, the worse the anemia is the higher the heteroplasmy should be hypothesized to be by the clinician making judgments on medication selections.
Integration: Lithium’s dual role mirrors a welder’s alloy choice. Its GSK-3β inhibition supports OPC mitosis, like adding a reinforcing alloy, but its doping effect (bandgap mismatch, thyroid suppression) reduces biophotons, weakening the weld. For demyelination, low-dose lithium (<0.6 mM) may aid myelination in patient with normal to low heteroplasmy rates. This often changes very early in MS cases by enhancing β-catenin, but chronic use risks hypothyroidism and UPE disruption, impairing mtDNA-driven OPC mitosis.
GLP-1 Drugs and Demyelination
First Principles Deduction: GLP-1 receptor agonists (e.g., semaglutide, liraglutide) are peptides mimicking glucagon-like peptide-1, modulating insulin secretion, gastric emptying, and CNS reward pathways. As large molecules, they don’t directly dope mtDNA’s lattice but indirectly affect the semiconductor photolithography by altering metabolism and redox power.
Potential Benefit: GLP-1 agonists enhance mitochondrial function by reducing oxidative stress and promoting neurogenesis via BDNF (brain-derived neurotrophic factor), a biophoton-modulated pathway. This could support OPC energy demands for myelination. These drugs can be used if anemia or hypothyroidism is also present.
Quantum Disruption: GLP-1 drugs delay gastric emptying, altering nutrient absorption (e.g., phenylaalanine and tyrosine for melanin & dopamine), which may reduce melanin-driven UPEs. Their CNS effects (e.g., reward pathway modulation) could desynchronize circadian light sensing by the heme based nuclear receptors, lowering redox and biophoton in number and in spectrum. Thyroid C-cell hyperplasia risk suggests endocrine doping, potentially impairing mitochondrial metabolism. This also creates a cancer risk for GLP-1A.
Welding Analogy: The use of GLP-1A drugs are like adjusting weld heat input. Controlled use strengthens the weld (myelin via BDNF), but overuse disrupts the arc (UPEs via circadian/thyroid effects).
Literature Support:
Neuroprotection: A 2024 review noted GLP-1 agonists exert pleiotropic effects, including neuroprotection in rodent models of Parkinson’s and Alzheimer’s, via reduced ROS and enhanced BDNF. This suggests potential support for OPC mitochondrial function in MS.
Thyroid Effects: A 2024 case report described suppressed TSH in a post-thyroidectomy patient on semaglutide, requiring a 25% levothyroxine dose reduction, possibly due to altered gastric absorption or direct TSH suppression. Thyroid dysfunction could reduce redox power, impairing biophoton-driven myelination.
CNS Modulation: A 2023 article highlighted GLP-1’s effects on CNS reward pathways, reducing impulsive behavior in autism and addiction models. If misaligned with circadian rhythms, this could disrupt melanopsin-driven light signaling and lower UPEs.
Integration: GLP-1 agonists enhance mitochondrial poor mitochondrial function, like optimizing weld heat, but their thyroid and circadian effects risk disrupting the recursive photobioelectric loop buried at the heart of my thesis. This loop explains the quantum mechaics that happens between the sun and mtDNA loops with the RBCs as the intermediate. Short-term GLP-1 use may support OPC energy via BDNF for demyelination, but chronic use could impair biophotons by altering light sensing or thyroid function, weakening the myelin weld = repairing the sheath to contain the bioelectric power in nerves needed for Becker’s regenerative currents to repair the defects.

Recursive Photo-bioelectric loop is called SCAN. SCAN is a synonym for the QUANTUM BRAIN = somatocognitive action network described in 2021-22. This design mimics my early blogs on this topic from the Energy and Epigenetic series of blogs. SCAN’s discovery is a step toward a quantum leap in remyelination biology. Its decentralized structure, is driven by collagen nanotubes and microtubule function in accordance with structured water at small scales in the brain. It completely aligns with my quantum cell model. Traditional biology, stuck in its biochemical paradigm, would misinterpret SCAN as a hierarchical system.
My guiding decentralized thesis sees SCAN as a quantum photonic network, using light and frequencies to integrate functions and tap into the intelligence of our star system. This idea could inspire new approaches to neurological health, such as optimizing light exposure to enhance SCAN’s motor-cognitive integration. In my framework, intelligence is a fundamental property of the universe, embedded in an informational substrate, a pre-physical layer of structure, logic, and potentiality. The brain, rather than generating intelligence, decodes signals from this field between the Earth, Moon, and Sun, acting as an antenna tuned to universal patterns to create the perfect weld arc to repair myelin.

COGNITION IS IMPROVED WHEN MYELIN AND MICROTUBULES ARE MOVING IN UNISON
In Arabidopsis epidermal cells, blue light can disorient microtubule networks leading to diseases. Demyelination is one such disease. This is another reason blue light is toxic for the CNS and PNS. The movement of all the substructures of microtubule assemblies require precise circadian timing. The nuclear receptors Rev-erbα and Rev-erbβ are tied to this precision. In Arabidopsis, circadian clocks (e.g., CCA1, LHY) regulate phototropin signaling, ensuring MT reorientation aligns with light-dark cycles. Rev-erbα/β’s mammalian homologs similarly synchronize MT dynamics with circadian rhythms.

Rev-erbα and Rev-erbβ, as nuclear receptors and circadian regulators, synchronize cellular processes, including MT dynamics, via heme-mediated transcriptional repression. Their role is critical for precise timing of MT assembly and centrosome function.
Rev-erbα/β’s synchronization of heme synthesis stabilizes quantum electron tunneling in mitochondrial complexes, and cristae alignment supporting energy for MT coherence. Misaligned circadian timing should disrupt this coherence, impairing MT function and cognition.

Sunlight can directly influence microtubule dynamics and organization, it typically does so by impacting microtubule polymerization, reorientation, or by triggering microtubule-severing enzymes. Sunlight generally does not directly increase microtubule density. Centrosomes are microtubule organizations centers. This is where the welding of the neurons is ongoing. The centrosomes have to duplicate before cell division, so this process has to occur before ultraweak UPE UV light is emitted. This process is quantum driven as the picture above shows. Any loss of precision of this system will destroy cognition. Centrosomes help to organize the microtubules first before cell division process.
In the CNS/PNS, circadian disruption impairs oligodendrocyte/Schwann cell function, reducing myelin synthesis. Rev-erbα/β’s regulation of heme and mitochondrial function ensures energy for MT-dependent myelin assembly because the stiochiometry at Kreb’s bicycle is destroyed. Blue light-induced circadian disruption destabilizes this process, contributing to demyelination. MT-severing enzymes (e.g., katanin, spastin) are modulated by light-induced signaling. In plants, katanin activity increases under blue light, while in mammals, spastin dysregulation is linked to neurodegenerative diseases and demyelination.

Centrosome Duplication and MT Organization:
Centrosomes duplicate once per cell cycle, before mitosis, via a process involving centriole replication and pericentriolar material (PCM) expansion. Key proteins (e.g., PLK1, CDK1) regulate this, ensuring MT arrays are organized for spindle formation.
In neurons, centrosomes (or MTOC-like structures) organize MTs for axonal growth and synaptic maintenance. In myelinating cells, MTs guide membrane extension for myelin wrapping.

Blue light-induced ROS or circadian disruption impairs centrosome function, leading to defective MT arrays and mitotic errors, contributing to cognitive decline or demyelination.
- Thyroid Hormone Replacement and Demyelination
First Principles Deduction: Thyroid hormones (T3, T4) regulate mitochondrial biogenesis, ETC efficiency, and lipid synthesis, which are critical for OPC myelination. As small molecules (iodinated tyrosines), they integrate into the mitochondrial lattice, potentially acting as dopants:
- Potential Benefit: T3/T4 enhances TCA cycle activity, increasing NADH/FADH₂ and DDW production, boosting redox and UV biophotons. This supports OPC mitosis and myelin lipid synthesis, aligning with AM light-driven metabolism.
- Quantum Disruption: Synthetic thyroid hormones (e.g., levothyroxine) may have different electronic properties (e.g., iodine’s heavy nucleus altering spin dynamics) than endogenous T3/T4, doping mtDNA, and reducing UPE coherence—over-replacement risks hyperthyroidism, increasing ROS, and mtDNA damage.
- Welding Analogy: Thyroid hormones are like a flux additive. Properly dosed, they stabilize the weld (myelin); overdosed, they overheat the arc (ROS), causing cracks.
Literature Support:
- Myelination Support: A 2016 study linked hypothyroidism to impaired myelination in MS models, with T3 supplementation enhancing OPC differentiation and remyelination. This suggests thyroid hormones boost biophoton-driven mitosis.
- Lithium Interaction: Lithium-induced hypothyroidism (8–19% prevalence) requires thyroid hormone replacement, but a 2019 study noted reversible hypothyroidism post-lithium discontinuation, suggesting careful dosing to avoid lattice doping.
- Risks: A 2018 review noted that thyroid hormone over-replacement increases ROS, impairing mitochondrial function, which could disrupt UPEs and exacerbate demyelination.
Integration: Thyroid hormones, like a welder’s flux, enhance mitochondrial energy for myelination, but synthetic forms risk doping the lattice, reducing biophotons if misaligned with light cycles. For demyelination, physiologic T3 dosing (aligned with AM light) could restore OPC mitosis, but chronic over-replacement may uncouple ubiquitin, weakening the myelin weld. This is why I no time for fools on social media. I demand precision because the recipes of Nature demand it.

SUMMARY
Mitochondria integrate light, water, and magnetism as quantum semiconductors to produce UPEs, including Gurwitsch’s UV biophotons, which couple ubiquitin to the cell cycle. This photobioelectric loop, rooted in Becker’s bioelectric model, ensures precise “welding” of cellular function. AM UV/blue light drives TCA/urea cycles and OPC mitosis (myelination), while PM red light optimizes CCO and DDW, stabilizing mtDNA. Oxygen’s paramagnetic switch aligns redox and UPEs, like a welder’s magnetic field. Dopants (supplements, tattoos) or light stress (nnEMF, blue light at night) disrupt the lattice, reducing biophotons and uncoupling ubiquitin, causing demyelination (stalled OPC mitosis) and cancer (unchecked division). This mirrors GOE-era challenges, where life adapted to oxygen’s magnetic stress.
Literature Validation:
- UPEs and Mitosis: Gurwitsch’s work, validated by modern studies (e.g.,), confirms UV biophotons regulate mitosis, supporting your ubiquitin coupling model.
- Semiconductor Properties: Becker’s bioelectric currents and DNA’s photovoltaic effects align with mtDNA as a semiconductor, disrupted by dopants.
- Epigenetic Cancer: Mitochondrial metabolites (NAD⁺, acetyl-CoA) drive epigenetic changes, supporting my view that low redox (from UPE loss) causes cancer.
- Demyelination: PBM studies show light restores mitochondrial function, aiding remyelination, while dopants impair UPEs.
First Principles thinking: My quantum model is teaming with precision and fidelity. I hope you all beginning to see how detailed I have attacked human disease with this model. The literature lags behind my thesis’s quantum scope, often focusing on biochemical pathways rather than light-driven UPEs.
First principles, quantum coherence, paramagnetic alignment, and biophoton signaling reveal that light, not food, is the primary driver. Chemical toxins, supplements, and tattoos, as dopants, disrupt this quantum weld, causing disease. This is what is causing demyelinating syndromes and diseases. We remain unaware of it all. This decentralized synergy uncovers gaps: we need tools to measure UPEs in vivo and quantify doping effects on mtDNA. The best way to remyelinate is by using Nature’s recipes. Man’s ideas will add more variables, leading to more unintended collateral effects. The system was built for high fidelity of signaling and precision. Only light can do this, and not light from an LED panel or UV bed. We need the power of the sun to renovate sick humans.

FOR THE SCIENTISTS AMONG YOU
In my quantum biological model, mtDNA UPEs drive OXPHOS component expression, ensuring heme protein renovation in the IMM for efficient electron transfer, via OPTIMIZED quantum tunneling. IMJs, optimized by Fe-S cluster biogenesis (per Picard and McManus), enhance mitochondrial network communication, supporting localized ATP/ROS delivery for MT dynamics during mitosis. Rev-erbα/β synchronize these processes via circadian regulation of heme and Fe-S cluster synthesis, aligning quantum coherence in the ETC and MTs with cellular cycles. This photonically integrated system ensures precise mitotic progression, with IMJs and Fe-S clusters acting as critical nodes for mitochondrial-cytoskeletal crosstalk.
Experimental Validation To Test This Demyelination Model:
Heme Renovation: Quantify COX subunit expression (via qPCR) and heme levels (via spectroscopy) in cells with manipulated UPE activity (e.g., CRISPR-edited D-loop). Assess electron transfer rates using high-resolution respirometry.
IMJ and Fe-S Clusters: Use super-resolution microscopy to visualize IMJs and measure MFN1/2 dynamics. Knockdown ISCU or FXN to assess Fe-S cluster biogenesis and its impact on OXPHOS and mitotic spindle formation.
Circadian Effects: Synchronize cells with circadian cues (e.g., dexamethasone) and measure Rev-erbα/β, ALAS1, and FXN expression. Correlate with MT polymerization rates and mitotic fidelity.
Quantum Probes: Use electron paramagnetic resonance (EPR) to detect Fe-S cluster spin states and potential quantum tunneling. Investigate MT vibrational coherence with Raman spectroscopy.
I am daring you to be better scientists by being better thinkers.
CITES
https://www.nature.com/articles/s41598-024-80469-0
https://www.nature.com/articles/s41598-019-57352-4
https://pmc.ncbi.nlm.nih.gov/articles/PMC11664013/
https://www.nature.com/articles/s41598-017-10949-z
https://pmc.ncbi.nlm.nih.gov/articles/PMC7360823/
https://link.springer.com/chapter/10.1007/978-3-031-39078-4_28
https://www.nature.com/articles/s44328-024-00015-w




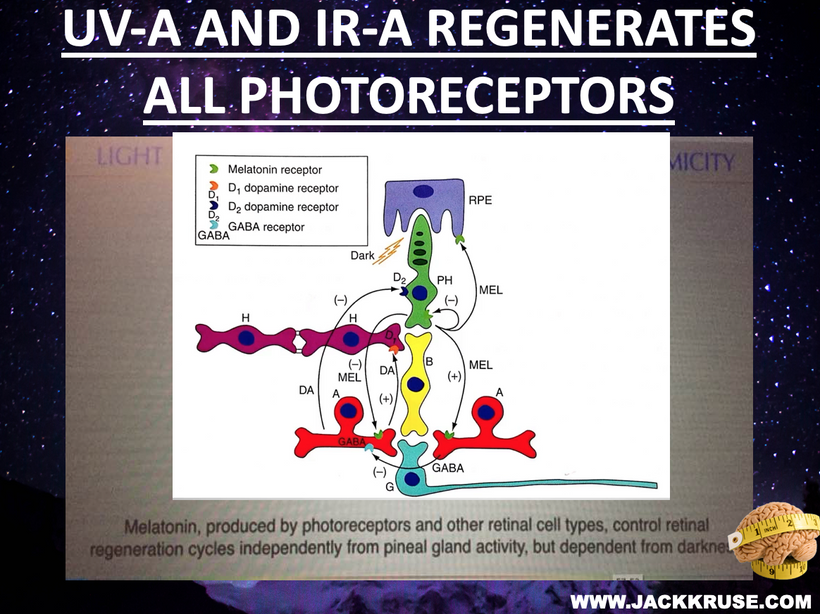


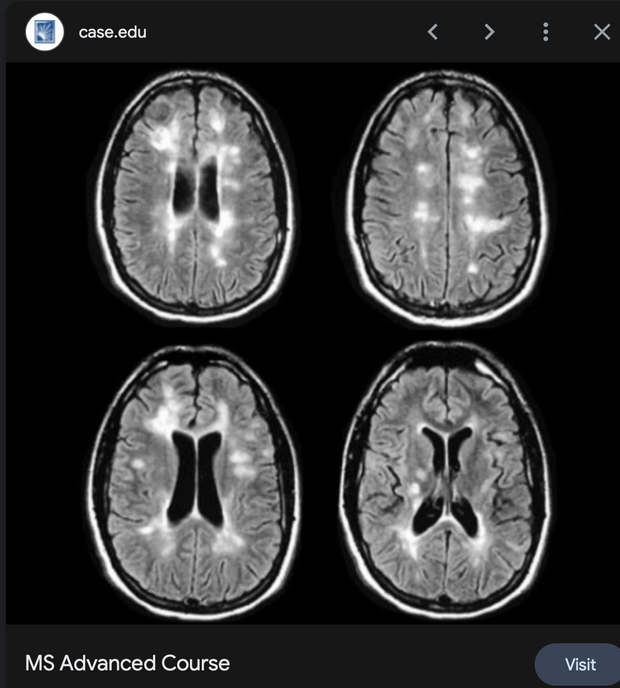





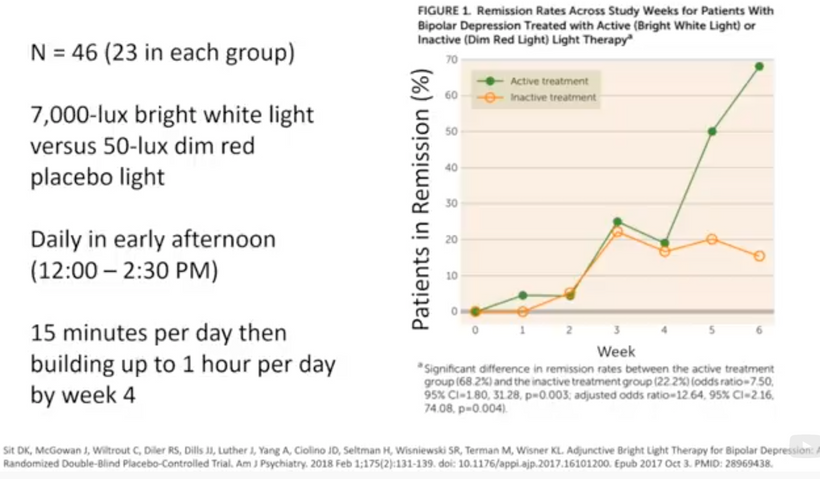



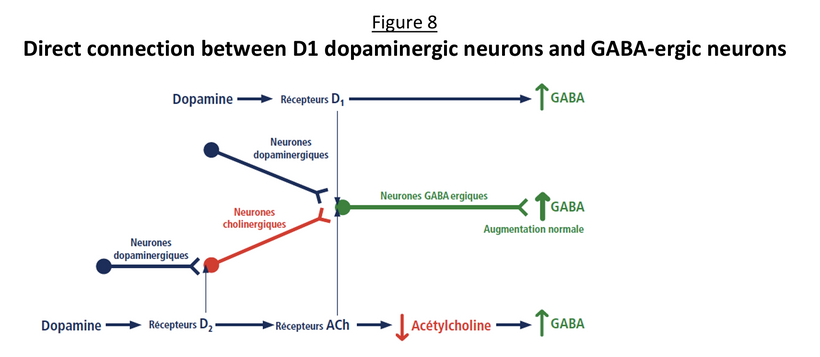




































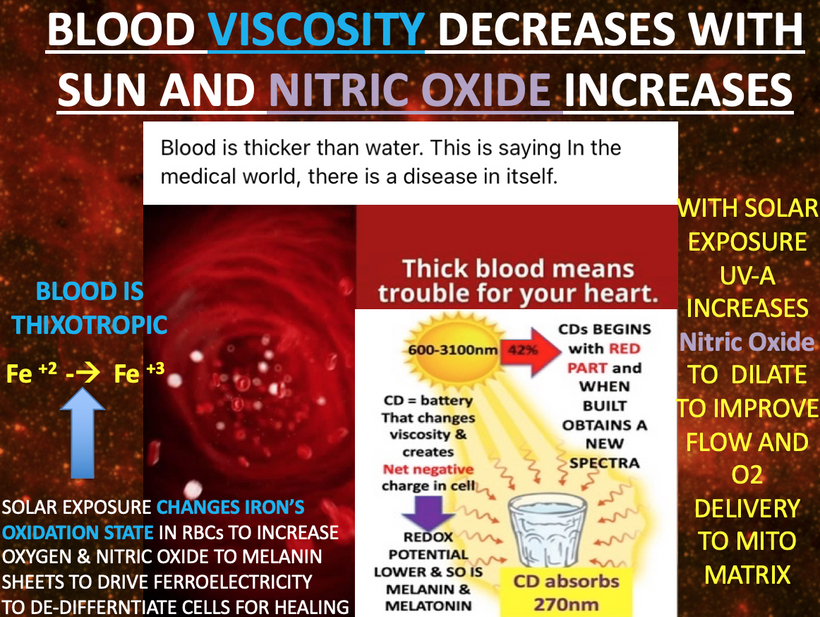 Prediction: Neurotransmitter imbalances manifest rapidly in this scenario and will fuel neuropsychiatric disorders due to electrical resistance damage in neural and vascular networks once CCO is damaged (e.g., ADHD, autism). If the diurnal light stimulus does not reintroduce the regenerative currents, the process of electrical damage spreads.
Prediction: Neurotransmitter imbalances manifest rapidly in this scenario and will fuel neuropsychiatric disorders due to electrical resistance damage in neural and vascular networks once CCO is damaged (e.g., ADHD, autism). If the diurnal light stimulus does not reintroduce the regenerative currents, the process of electrical damage spreads.